Электрохимические методы анализа - это совокупность методов качественного и количественного анализа, основанных на электрохимических явлениях, происходящих в исследуемой среде или на границе раздела фаз и связанных с изменением структуры, химического состава или концентрации анализируемого вещества.
Разновидностями метода являются электрогравиметрический анализ (электроанализ), внутренний электролиз, контактный обмен металлов (цементация), полярографический анализ, кулонометрия и др. В частности, электрогравиметрический анализ основан на взвешивании вещества, выделяющемся на одном из электродов. Метод позволяет не только проводить количественные определения меди, никеля, свинца и др., но и разделять смеси веществ.
Кроме того, к электрохимическим методам анализа относят методы, основанные на измерении электропроводности (кондуктометрия) или потенциала электрода (потенциометрия). Некоторые электрохимические методы применяются для нахождения конечной точки титрования (амперометрическое титрование, кондуктометрическое титрование, потенциометрическое титрование, кулонометрическое титрование).
Различают прямые и косвенные электрохимические методы. В прямых методах используют зависимость силы тока (потенциала и т.д.) от концентрации определяемого компонента. В косвенных методах силу тока (потенциал и т. д.) измеряют с целью нахождения конечной точки титрования определяемого компонента подходящим титрантом, т.е. используют зависимость измеряемого параметра от объема титранта.
Для любого рода электрохимических измерений необходима электрохимическая цепь или электрохимическая ячейка, составной частью которой является анализируемый раствор.
Электрохимические методы классифицируют в зависимости от типа явлений, замеряемых в процессе анализа. Различают две группы электрохимических методов:
1. Методы без наложения постороннего потенциала, основанные на измерении разности потенциалов, который возникает в электрохимической ячейке, состоящей из электрода и сосуда с исследуемым раствором. Эту группу методов называют потенциометрическими. В потенциометрических методах используют зависимость равновесного потенциала электродов от концентрации ионов, участвующих в электрохимической реакции на электродах.
2. Методы с наложением постороннего потенциала, основанные на измерении: а) электрической проводимости растворов - кондуктометрия ; б) количества электричества, прошедшего через раствор - кулонометрия ; в) зависимости величины тока от приложенного потенциала - вольт-амперометрия ; г) времени, необходимого для прохождения электрохимической реакции - хроноэлектрохимические методы (хроновольтамперометрия, хронокондуктометрия). В методах этой группы на электроды электрохимической ячейки налагают посторонний потенциал.
Основным элементом приборов для электрохимического анализа является электрохимическая ячейка. В методах без наложения постороннего потенциала она представляет собой гальванический элемент , в котором вследствие протекания химических окислительно-восстановительных реакций возникает электрический ток. В ячейке типа гальванического элемента в контакте с анализируемым раствором находятся два электрода - индикаторный электрод, потенциал которого зависит от концентрации вещества, и электрод с постоянным потенциалом - электрод сравнения, относительно которого измеряют потенциал индикаторного электрода. Измерение разности потенциалов производят специальными приборами - потенциометрами.
В методах с наложением постороннего потенциала применяют электрохимическую ячейку , названную так потому, что на электродах ячейки под действием наложенного потенциала происходит электролиз - окисление или восстановление вещества. В кондуктометрическом анализе используют кондуктометрическую ячейку, в которой замеряют электрическую проводимость раствора. По способу применения электрохимические методы можно классифицировать на прямые, в которых концентрацию веществ измеряют по показанию прибора, и электрохимическое титрование, где индикацию точки эквивалентности фиксируют с помощью электрохимических измерений. В соответствии с этой классификацией различают потенциометрию и потенциометрическое титрование, кондуктометрию и кондуктометрическое титрование и т.д.
Приборы для электрохимических определений кроме электрохимической ячейки, мешалки, нагрузочного сопротивления включают устройства для измерения разности потенциалов, тока, сопротивление раствора, количества электричества. Эти измерения могут осуществляться стрелочными приборами (вольтметр или микроамперметр), осциллографами, автоматическими самопишущими потенциометрами. Если электрический сигнал от ячейки очень слабый, то его усиливают с помощью радиотехнических усилителей. В приборах методов с наложением постороннего потенциала важной частью являются устройства для подачи на ячейку соответствующего потенциала стабилизированного постоянного или переменного тока (зависит от типа метода). Блок электропитания приборов электрохимического анализа включает обычно выпрямитель и стабилизатор напряжения, который обеспечивает постоянство работы прибора.
Потенциометрия объединяет методы, основанные на измерении эдс обратимых электрохимических цепей, когда потенциал рабочего электрода близок к равновесному значению.
Вольтамперометрия основана на исследовании зависимости тока поляризации от напряжения, прикладываемого к электрохимической ячейке, когда потенциал рабочего электрода значительно отличается от равновесного значения. Широко используется для определения веществ в растворах и расплавах (например, полярография, амперометрия).
Кулонометрия объединяет методы анализа, основанные на измерении количества вещества, выделяющегося на электроде в процессе электрохимической реакции в соответствии с законами Фарадея . При кулонометрии потенциал рабочего электрода отличается от равновесного значения.
Кондуктометрический анализ основан на изменении концентрации вещества или химического состава среды в межэлектродном пространстве; он не связан с потенциалом электрода, который обычно близок к равновесному значению.
Диэлектрометрия объединяет методы анализа, основанные на измерении диэлектрической проницаемости вещества, обусловленной ориентацией в электрическом поле частиц (молекул, ионов), обладающих дипольным моментом. Диэлектрометрическое титрование используют для анализа растворов.
Электрохимические методы – наиболее динамично развивающиеся с точки зрения их применения в экологическом мониторинге. Наиболее часто в системах МОС используют вольтамперометрию (включая полярографию), потенциометрию (в т.ч. ионометрию), кулонометрию и кондуктометрию.
Электрохимические методы анализа используют зависимость различных электрических свойств среды от количественного содержания и качественного состава анализируемых в ней веществ:
· изменение потенциала электрода в зависимости от физико-химических процессов, протекающих в веществе (потенциометрический метод), в т.ч. селективные реакции ионоселективных электродов, индивидуально чувствительных к большому числу катионов и анионов (ионометрический метод);
· изменение электропроводности (тока) и диэлектрической проницаемости вещества в зависимости от природы среды и концентрации ее компонентов (кондуктометрический и амперометрический методы);
· изменения количества электричества при попадании определяемого вещества в электрохимическую ячейку (кулонометрический метод);
· восстановление анализируемого соединения на ртутном капающем или вращающемся электроде, как правило, при анализе следовых количеств веществ, находящихся в разных агрегатных состояниях (полярографический или вольтамперометрический метод).
Полярографы из всех приборов этой группы имеют наивысшую чувствительность, равную 0,005–1 мкг/мл пробы.
Вольтамперометрия включает в себя группу электрохимических методов анализа, основанных на изучении поляризационных кривых. Эти методы – полярография и амперометрическое титрование – имеют множество разновидностей и модификаций. Наиболее распространена постоянно-токовая полярография .
Полярографическая установка состоит из источника постоянного тока, делителя напряжения, капельного (обычно ртутного) или вращающегося электрода и вспомогательного (обычно тоже ртутного или другого) электрода. Дляизмерения силы тока в систему подключают микроамперметр. Электроды помещены вместе с исследуемым раствором в электролизер (ячейку).
Наложенное на электролитическую ячейку напряжение вызывает поляризацию анода и катода E = f a – f k +iR , где i – сила тока; К – сопротивление раствора; f a и f k – потенциалы анода и катода.
Если уменьшить сопротивление раствора, добавив сильный электролит (фон), то величиной iR (падение потенциала в растворе) можно пренебречь.
Потенциал анода практически остается постоянным во время работы электролизера, таккак плотность тока мала и относительно большая поверхность анода не поляризуется. Тогда потенциал капающего поляризующего катода с небольшой поверхностью будет равен: Е = -f k . Часто в полярографических измерениях вместо слоя ртути на дне сосуда применяют неполяризующийся насыщенный каломелевый электрод, потенциал которого принимают равным нулю.
Полярографические данные получают путем измерения тока, проходящего через электролитическую ячейку, как функции потенциала, налагаемого на электроды. Графическую зависимость силы тока от потенциала называют полярографической волной (рис. 2 ).
В начале электролиза при небольших значениях наложенной ЭДС сила тока будет почти постоянной и лишь очень медленно возрастать. Это так называемый остаточный ток, который сохраняется во все время электролиза.
Рис. 2 . Полярограмма 10 –3 М раствора хлорида цинка и 1 М раствора хлорида калия (кривая 1) и 1 М раствора хлорида калия (кривая 2)
Как только будет достигнут потенциал восстановления ионов (например, для определяемых ионов цинка он равен -1,0 В), начинается их разряд на капле ртути:
Zn 2+ + 2 +Hg ® Zn (Hg).
На катоде образуется разбавленная амальгама цинка Zn (Hg), которая разлагается насвои составляющие, как только падающая капля соприкоснется с анодом:
Zn (Hg) – 2 ® Zn 2+ +Hg.
При потенциале восстановления ионов цинка сила тока резко возрастает (рис. 2 ), но после достижения определенной величины, несмотря на увеличение приложенной ЭДС, она остается почти постоянной. Этот ток называется предельным или диффузионным, его величина,как правило, пропорциональна концентрации определяемого вещества.
При снятии полярограмм к исследуемому электролиту добавляют индифферентный электролит с катионами, восстанавливающимися гораздо труднее анализируемого катиона, например, КСl, KNO 3 , NH 4 Cl; при концентрации в 100–1000 раз превышающей концентрацию определяемого вещества. Такой электролит называют «фоновым». Его создают в исследуемом растворе для увеличения электропроводности и для экранирования электрического поля индикаторного электрода (катода). Поэтому катионы определяемого вещества не притягиваются электрическим полем катода, а двигаются к нему за счет диффузии.
Важнейшей характеристикой полярограммы является потенциал полуволны Е 1/2 и высота полярографической волны h (предельный диффузионный ток). Потенциал полуволны используют в качественном полярографическом анализе. Потенциалы полуволны различных веществ, расположенные в порядке возрастанияих отрицательного значения, составляют так называемый «полярографический спектр». Поскольку потенциал полуволны существенно зависит от состава раствора (анализируемой среды), в полярографических таблицах всегда указывается фон.
В количественном полярографическом анализе для измерения концентрации используют методы градуировочного графика, добавок, сравнения и расчетный метод.
Среди различных вариантов полярографии метод дифференциальной импульсной полярографии (ДИП ) наиболее эффективен для решения задач экологического мониторинга, главным образом благодаря высокой чувствительности. Метод ДИП позволяет оценивать содержание всех веществ, определяемых методом классической полярографии. Среди других полярографических методов, особенно удобна для следового анализа квадратно-волновая полярография , которая обеспечивает предел обнаружения, близкий к пределу обнаружения ДИП, но только в случае обратимых электродных процессов, и поэтому этот метод часто используется для определения следов тяжелых металлов. Метод ДИП может использоваться и для определения поверхностно-активных веществ, изменяющих емкость двойного электрического слоя электрода.
Для определения микросодержаний ионов тяжелых металлов могут быть использованы методы инверсионною электрохимического анализа (ИЭА) или по-другому, инверсионного вольтамперометрического анализа (ИВА ), в которых определяемые металлы предварительно осаждают на электроде и затем растворяют при полярографическом контроле. Этот вариант в сочетании с ДИП относится к наиболее чувствительным методам электрохимического анализа. Аппаратурное оформление ИЭА (ИВА) относительно несложное, что позволяет проводить анализы в полевых условиях, причем автоматизированные станции непрерывного контроля (мониторинга) также могут работать на этом принципе.
Методы ИЭА (ИВА) обеспечивают определение ионов Сu, РЬ, Bi, Sb, As, Sn In, Ga, Ag, Tl, Cd, Zn, Hg, Аu, Ge, Те, Ni, Со и многих анионов. Важным преимуществом методов ИЭА (ИВА) является (в отличие от других методов, например, таких, как атомно-абсорбционная спектрометрия) способность отличать свободные ионы oт их связанных химических форм , что важно и для оценки физико-химических свойств анализируемых веществ с точки зрения экоаналитического контроля (например, при оценке качества воды). Многие органические вещества также могут быть определены методами ИЭА (ИВА) после их адсорбционного накопления на поверхности электрода.
Полярографическими методами можно также определять аэрозоли различных металлов в атмосфере и воздухе производственных помещений после их улавливания на соответствующих фильтрах с последующим переведением концентратов в раствор. Органические соединения, находящиеся в виде газов и паров в атмосфере, могут быть определены полярографически после их поглощения специально подобранными растворами. Металлы и различные соединения в биологических материалах обычно определяют полярографически после их экстракции. Все полярографические измерения, в т. ч. ИЭА (ИВА), могут быть полностью автоматизированы, что существенно при выполнении серийных анализов.
Одной из важнейших областей применения полярографии является определение кислорода в воде. Для этого используют амперометрические детекторы, генерирующие ток, пропорциональный концентрации кислорода в растворе.
Нанося фермент на поверхность мембраны детектора можно получать различные ферментные амперометрические сенсоры, удобные для биохимических и клинических анализов. Такие сенсоры применяют и в системах экологического мониторинга .
Электроды, работающие по электрокаталитическому принципу , пригодны для мониторинга различных газов (SО 2 , H 2 S, CO, NO x) в воздухе производственных помещений. Электрохимические реакции этих газов (играют роль катализатора), протекающие на поверхности электрода, генерируют в электродной системе ток, функционально связанный с концентрацией газов в воздухе.
Применение полярографии не ограничивается анализом дискретных проб, и метод постепенно переходит на принципы непрерывного анализа газов и жидкостей.
Вольтамперометрические полярографические детекторы успешно применяются в высокоэффективной жидкостной хроматографии (ВЭЖХ). В этом случае сочетание высокоселективного способа разделения с чувствительным способом детектирования приводит к заметному расширению номенклатуры веществ, определяемых хроматографическим методом (следы высокотоксичных веществ, гербициды, лекарственные препараты, стимуляторы роста и др.).
Подробности метода можно уточнить в специальной литературе ,,,,.
Потенциометрия – метод определения концентрации веществ, основанный на измерении ЭДС обратимых гальванических элементов.
На практике используют два аналитических метода: прямую потенциометрию для определения активности частиц, которую можно рассчитать с помощью уравнения Нернста по ЭДС гальванического элемента, и потенциометрическое титрование , в котором изменение активностей химических веществ в процессе титрования приводит к изменению ЭДС гальванического элемента.
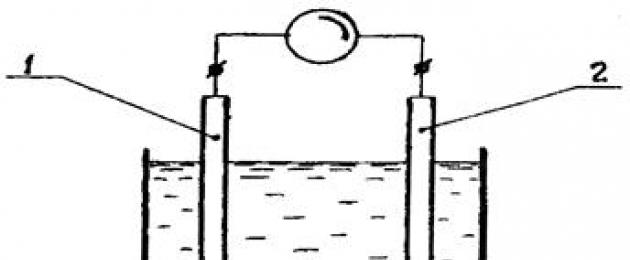
Аппаратура для проведения потенциометрических титрований и для прямой потенциометрии одна и та же. В схему потенциометрических измерений входят индикаторный электрод и электрод сравнения, обладающий устойчивым постоянным потенциалом, в также вторичный прибор. Принципиальная схема метода показана на рис. 3 .

1 – индикаторный электрод; 2 - электрод сравнения
Рис. 3. Потенциометрическая ячейка
Потенциал пары электродов постоянен. Изменение концентрации анализируемого вещества в растворе изменяет ЭДС цепи. Индикаторные электроды обычно бывают четырех типов , в зависимости от применяемой мембраны которая отделяет раствор электрода от исследуемого раствора: 1) электроды с гомогенной мембраной из порошкообразного или кристаллического материала; 2) электроды с гетерогенной мембраной, в которых электродно активное вещество распределено, например, в силиконовой резине; 3) электроды с жидкой мембраной, в которых мембрана – это раствор, нанесенный на нейтральное вещество, например, пористое стекло; 4) стеклянные электроды с различным химическим составом стекла.
Индикаторные электроды приобретают потенциал раствора, в который они помещены. Различают два вида индикаторных электродов:
1) электроды индифферентные (неразрушаемые в ходе электролиза);
2) электроды изменяющиеся (окисляющиеся или восстанавливающиеся) во время измерений.
Роль индифферентных электродов (их иногда называют электродами третьего рода) заключается в том, чтобы отдавать или присоединять электроны, т.е. быть проводниками электричества. Такие электроды могут быть изготовлены из золота, полированной платины, графита других материалов. Примерами изменяющихся электродов (иногда их называют электродами первого рода) могут быть пластины из меди, цинка и других металлов, а также хингидронный и водородный индикаторный электроды. Индикаторными электродами могут быть, кроме того, ионселективные мембранные электроды для определения многочисленных катионов: Li + , Рb + , Cs + , Тl + , NH + , Na + , К + , Аg + и др. В качестве электродов сравнения (стандартные электроды ), потенциал которых остается постоянным на протяжении измерения, чаще всего используется, например, нормальный и децинормальный каломелевые (каломельные) электроды с потенциалами +0,282 В и +0,334 В, соответственно, а также насыщенный хлорсеребряный электрод с потенциалом +0,201 В.
В идеальном случае прямое потенциометрическое измерение ЭДС гальванического элемента может быть связано через уравнение Нернста с активностью определяемой частицы, либо с концентрацией, если известны соответствующие коэффициенты активности:
![]()
где Е 0 – стандартный потенциал электрода, В; R – газовая постоянная; T – абсолютная температура; F – число Фарадея; n – число теряемых или получаемых электронов; , [восст.] – равновесные концентрации окисленной, восстановленной форм соответственно, моль/дм 3 .
Если подставить эталонные значения констант и перейти от натурального логарифма к десятичному, то для температуры 25°С получим;

Важнейшим показателем при характеристике состоянии ОС является значение рН этой среды, определение которого (рН–метрия ) в настоящее время обычно проводят с помощью стеклянных индикаторных (измерительных) электродов. Для долговременных измерений разработаны специальные конструкции стеклянных электродов с дополнительными устройствами, обеспечивающими очистку стеклянной мембраны. Стеклянные электроды, покрытые полупроницаемой мембраной с пленкой электролита, служат также основой различных типов зондов (сенсоров ), применяемых в анализе вод и воздуха в условиях производства на ряд загрязнений (NH 3 , СО 2 , NO x ,SО 2 , H 2 S и др.).
Процесс в области создания ион-селективных электродов (ИСЭ) позволяет осуществлять контроль ионов F – , I – , Br – , Cl – , CN – , SCN – , NO 3 – , NO 2 – , ClO 4 – , S 2– , Na + , К + Са 2+ , Аg + , Си 2+ , Cd 2+ , РЬ 2+ в интервалах концентрации от 10 –2 до 10 –7 моль/л (примерно 1– 10 –5 мг/мл). Контроль с помощью ИСЭ отличается экспрессностью, простотой и большими возможностями проведения непрерывных измерений. Разработаны ИСЭ, селективные к широкому классу органических веществ, а также изомеров в их массе, поверхностно-активных и моющих веществ, находящихся в воздухе производственной зоны и водно-хозяйственного режима промышленных предприятий.
Потенциометрию используют и при измерениях окислительно-восстановительных потенциалов различных окислительно-восстановительных (О/В) систем в воде. Как правило, результаты измерения соответствуют смешанному потенциалу, так как обычно в воде одновременно сосуществуют несколько О/В систем.
Следует отметить перспективность использования сенсоров на основе полупроводниковых металлоксидных химически селективных и ион-селективных полевых транзисторов (ХСПТ, ИСПТ). Селективность в этих системах достигается выбором состава мембраны и слоя, осажденного на затвор транзистора. Систему погружают в анализируемый раствор, и разностью потенциалов между электродом сравнения и затвором транзистора модулируют ток, протекающий между его истоком и стоком. Вследствие селективности мембраны или осажденного слоя, модулированный ток становится функцией активности соответствующего компонента раствора. Полупроводниковые сенсоры составляют основу мониторов–анализаторов различных газов и паров. Малые размеры таких сенсоров позволяют объединять их совокупности в виде мозаики на единой подложке, так что получается анализатор, способный контролировать целый набор вредных веществ. Сигналы от отдельных сенсоров, входящих в мозаику могут последовательно и периодически регистрироваться измерительным центром аналитической системы.
Развитие микроэлектроники делает возможным конструирование компактных анализаторов типа зондов с использованием современных ИСЭ. При этом в ручке зонда может быть смонтирована схема, обрабатывающая отклик с объекта экологического контроля, и даже дисплей.
В специальной литературе можно ознакомиться с подробностями метода , , , .
Кулонометрический метод анализа представляет собой измерение тока электродной реакции, в которую вступает исследуемое вещество, попадающее в кулонометрическую ячейку с анализируемым потоком. Принципиальная схема кулонометрической ячейки показана на рис. 4 .

1 – катодная камера; 2 – анодная камера; 3 – микроамперметр
Рис. 4 . Схема кулонометрической ячейки
Кулонометрический анализ основан на измерении количества электричества, затраченного на количественное проведение данного электрохимического процесса в данной пробе, т.е. при условии, что выход по току равен 100%. Это количество электричества при помощи включенного в цепь последовательно с измерительной ячейкой интегратора ток-время, либо кулонометра-электролизера, в котором осуществляется электрохимический процесс со стопроцентным выходом по току, сопровождающийся выделением вещества, количество которого можно легко и точно восстановить.
В соответствии с законом Фарадея:
m(x )/M (x ) = m (k )/M (k ),
где m (x ), m(k) – массы определяемого вещества х и вещества, выделяемого в кулонометре, соответственно; M (x ), M (k ) – молярная масса эквивалентов вещества х и вещества, выделяемого в кулонометре, г/моль.

Расчет можно также производить по уравнению, описывающему закон Фарадея:
![]()
если при проведении анализа измеряют силу тока i , А и время t , с, затраченные на проведение электрохимического процесса.
В другой модификации данного метода, называемой
кулонометрическим титрованием
, титрант генерируют электролитически в анализируемом растворе при заданном токе. Потребление титранта в аналитической реакции восполняют зарядом, протекающим через раствор при генерировании титранта вплоть до достижения точки эквивалентности.
Одним из преимуществ кулонометрических методов является то, что процесс стандартизации титранта часто не является обязательным, так как расчеты основаны на постоянной Фарадея, т.е. метод является абсолютным и позволяет оценивать количество определяемого вещества, а не его концентрацию . Недостатком кулонометрии с заданным потенциалом является длительность процедуры анализа, связанная с необходимостью полного завершения электролиза. Вычислительная техника дает возможность сократить это время, предсказывая момент конца электролиза путем математической обработки кривой «ток–время» для начальных стадий электролиза и путем расчета количества электричества или концентрации вещества в растворе. При анализе многокомпонентных проб может быть использована сканирующая кулонометрия , в которой потенциал электролиза изменяют непрерывно или ступенчато. Для таких систем кулонометрическое титрование предпочтительнее прямой кулонометрии, так как 100%-ную эффективность тока при генерировании титранта достаточно просто достичь правильным выбором титрант–реагента и состава рабочей среды. Кулонометрическое титрование применимо для определения от 0,01 до 100 мг веществ (иногда ниже 1 мкг). Рабочий объем проб обычно составляет от 10 до 50 мл. Метод характеризуется высокой точностью, относительная погрешность не превышает нескольких десятых долей % даже при кулонометрическом титровании микрограммовых содержаний. В оптимальных условиях титрование может быть выполнено с очень малой суммарной погрешностью на уровне 0,01% (отн.). Различные кислотно-основные, окислительно-восстановительные; осади-тельные и комплексонометрические варианты титрования можно проводить кулонометрически.
Разработаны и выпускаются кулонометрические газоанализаторы и аква-анализаторы («кулонометры») для определения диоксида серы и сероводорода (сульфатов и сульфидов), озона (и перекиси водорода), хлора в воздухе (и активного хлора в воде), оксида углерода и диоксида азота в воздухе (нитратов и нитритов в воде). Кулонометрия используется также как средство электрохимического детектирования в жидкостной хроматографии.
С подробностями метода можно познакомиться в специальной литературе .
Кондуктометрический метод анализа основан на измерении электропроводности раствора. Кондуктометрический метод анализа заключается в измерении изменения сопротивления раствора электролита при поглощении компонента смеси. Кондуктометрические установки применяются, например, для определения оксида и диоксида углерода, паров бензина, аммиака и других.
Электропроводностью называют величину обратную сопротивлению R , ее размерность См (сименс) т.е. æ = 1/R .
Электропроводность раствора зависит от числа ионов в единице объема раствора, т.е. от концентрации С , от подвижности этих ионов – V. На основании известных соотношений
![]()
где Z – расстояние между электродами; S – площадь электродов; k –коэффициент пропорциональности.
Для конкретной пары электродов при неизменном расстоянии между ними S /Z = const. Тогда
![]() ,
,
где k 1 = k (S /Z ).
При расчетах в кондуктометрии используют понятие «удельная электрическая проводимость» æ 0:
![]()
В расчетах удобно пользоваться эквивалентной электропроводностью, которая равна:
где п – число молей эквивалента в 1 см 3 раствора. Эквивалентная электропроводность l ¥ при бесконечном разбавлении равна сумме подвижностей катиона U и аниона V.
Отношение эквивалентной электропроводности раствора слабого электролита к эквивалентной электропроводности этого электролита при бесконечном разбавлении равно степени диссоциации a этого электролита:
Несмотря на неспецифичность, этот метод довольно часто, по сравнению с другими электрохимическими методами, используются в системах экологического мониторинга. Это объясняется тем, что при оценке загрязненности, например, воды и атмосферы, возможен не постадийный, а выходной (конечный) контроль промышленных процессов. Из-за крайне низкой электропроводности воды чаще всего вполне достаточно оценить общее содержание загрязнений, что и обеспечивает кондуктометрия. Типичными примерами использования кондуктометрических методов в контроле окружающей среды являются анализаторы детергентов в сточных водах, концентрации синтетических компонентов в оросительных системах, качества (солености) питьевой воды. Кондуктометрические анализаторы используются для непрерывного контроля загрязнений воздуха и атмосферных осадков, например SO 2 и H 2 SO 4 . В дополнение к прямой кондуктометрии дляопределения некоторых видов загрязнения могут быть использованы косвенные методы, что обеспечивает весьма эффективные оценки содержания перечисленных выше веществ, которые взаимодействуют перед измерением со специально подобранными реагентами и регистрируемоеизменение электропроводности вызывается только присутствием соответствующих продуктов в реакции. Так можно определять оксиды азота послеих каталитического восстановления доаммиака, а также НСl, НВг и СО 2 после предварительной реакции с Ва(ОН) 2 или NaOH. Описанный принцип определении СО 2 может быть использован и для косвенного определения органических веществ в воде.
В дополнение к классической кондуктометрии имеется иее высокочастотный вариант (осциллометрия ), в котором индикаторная электродная система не контактирует с пробой. Этот принцип часто реализуется в кондуктометрических анализаторах непрерывного действия.
Электрохимические методы анализа также описаны еще в целом ряде учебных и специальных изданий , , , .
ЛИТЕРАТУРА
1. Другов Ю.С., Родин А.А.
Экологическая аналитическая химия.
С.-Петербург: 2002. – 464 с.
2. Пашкевич М.А., Шуйский В.Ф. Экологический мониторинг. Учебное пособие. СПбГГУ. – СПб., 2002. – 90 с.
3. Каттралл Роберт В. Химические сенсоры. М.: Научный мир, 2000. – 144 с.
4. Турьян Я.И., Рувинский О.Е., Зайцев П.М. Полярографическая каталиметрия. М.: Химия, 1998. – 272 с.
5. Будников Г.К., Майстренко В.Н., Муринов Ю.И. Вольтамперометрия с модифицированными и ультрамикроэлектродами. М.: Наука,1994. – 239с.
6. Брайнина Х.З., Нейман Е.Я., Слепушкин В.В. Инверсионные электроаналитические методы. М.: 1988. – 240 с.
7. Салихджанова Р.Ф. и др. Полярографы и их эксплуатация в практическом анализе и исследованиях. М.: Химия, 1988. – 192 с.
8. Каплан Б.Я., Пац Р.Г., Салихджанова Р.Ф. Вольтамперометрия переменного тока. М.: Химия, 1985. – 264.
9. Бонд А.М. Полярографические методы в аналитической химии. М.: Химия, 1983.
10. Ефременко О.А. Потенциометрический анализ. М.: ММА им. И.М. Сеченова, 1998.
11. Справочное руководство по применению ионселективных электродов. М.: Мир, 1986.
12. Корыта И. Ионы, электроды, мембраны. М.: Мир, 1983.
13. Никольский Б.В., Матерова Е.А. Ионселективные электроды. Л.: Химия, 1980.
14. Ефременко О.А. Кулонометрическое титрование. М.: ММА им. И.М. Сеченова, 1990.
15. Худякова Т.А., Корешков А.П. Кондуктометрический метод анализа. Учеб пособие для вузов. М.: Высшая школа, 1975. – 207 с.
16. Будников Г.К., Майстренко В.Н., Вяселев М.Р. Основы современного электроанализа. М.: Химия, 2000.
17. Прохорова Г.В. Введение в электрохимические методы анализа. М.: Изд-во МГУ, 1991. – 97 с.
18. Электроаналитические методы в контроле окружающей среды . /Под ред. Р. Кальвода, Р. Зыка, К. Штулик и др. М.: Химия, 1990. – 240 с.
19. Плэмбек Дж. Электрохимические методы анализа. Основы теории и применения. /Пер. с англ. М.: Мир, 1986.
Физико-химические методы анализа (ФХМА) основаны на использовании зависимости между измеряемыми физическими свойствами веществ и их качественным и количественным составом. Поскольку физические свойства веществ измеряются с помощью различных приборов – «инструментов», то эти методы анализа называют также инструментальными методами.
Наибольшее практическое применение среди ФХМА имеют:
- электрохимические методы – основаны на измерении потенциала, силы тока, количества электричества и других электрических параметров;
- спектральные и другие оптические методы – основаны на явлениях поглощения или испускания электромагнитного излучения (ЭМИ) атомами или молекулами вещества;
- хроматографические методы – основаны на сорбционных процессах, протекающих в динамических условиях при направленном перемещении подвижной фазы относительно неподвижной.
К достоинствам ФХМА можно отнести высокую чувствительность и низкий предел обнаружения – массовый до 10-9 мкг и концентрационный до 10-12 г/мл, высокую селективность (избирательность), позволяющую определять компоненты смесей без их предварительного разделения, а также экспрессность проведения анализов, возможность их автоматизации и компьютеризации.
В аналитической химии широко применяются электрохимические методы. Выбор метода анализа конкретного объекта анализа определяется многими факторами, в том числе, в первую очередь, нижним пределом определения элемента.
Данные о нижнем пределе обнаружения различных элементов некоторыми методами представлены в таблице.
Пределы определения (мкг/мл) элементов различными методами
| Элемент | МАС | ААС | ПТП | ИВА | Ионо- метрия | Ампером.титров. |
| Ag | 0,1– дитизон | 0,07 | 0,2 | 0.00001 | 0.02 | 0.05 |
| As | 0,05 - молибд.синь | 0,2 | 0,04 | 0,02 | - | 0,05 |
| Au | 0,04-метил.фиол. | 0,3 | 0,005 | 0,001 | - | 0,05 |
| Bi | 0,07-дитизон | 0,005 | 0,00001 | - | 0,5 | |
| Cd | 0,04-дитизон | 0,05 | 0,002 | 0,00001 | 0,03 | 0,5 |
| Cr | 0,04-дифе-нилкарбазид | 0,2 | 0,02 | - | - | |
| Cu | 0,03-дитизон | 0,2 | 0,002 | 0,00002 | 0,01 | 0,05 |
| Hg | 0,08-дитизон | - | 0,00005 | |||
| Pb | 0,08-дитизон | 0,6 | 0,003 | 0,00002 | 0,03 | |
| Sb | 0,08-родамин | 0,004 | 0,00004 | - | 0,5 | |
| Fe | 0,1-роданид | 0,2 | 0,003 | 0,0002 | 0,3 | 0,5 |
| Se | 0,08-диами-нофталин | 0,3 | 0,2 | 0,00002 | - | 0,5 |
| Sn | 0,07-фенил-флуриом | 0,4 | 0,003 | 0,00004 | - | 0,5 |
| Te | 0,1-висмутол | 0,7 | 0,02 | - | - | |
| Tl | 0,06-родамин | 0,6 | 0,01 | 0,00002 | - | 0,5 |
| Zn | 0,02-дитизон | 0,02 | 0,003 | 0,0003 | - | 0,5 |
| F - | - | - | - | - | 0,02 | 5-10 |
| NH 4 + ,NO 3 - | - | - | - | - | 0,1 | 1-5 |
МАС - молекулярная абсорбционная спекрометрия (фотометрия);
ААС - атомно-абсорбционная спектрометрия (пламенная фотометрия);
ПТП - переменно-токовая полярография;
ИВА - инверсионная вольтамперометрия.
Погрешности определений в ФХМА составляют около 2-5%, проведение анализов требует применения сложной и дорогостоящей аппаратуры.
Различают прямые и косвенные методы физико-химического анализа. В прямых методах используют зависимость величины измеряемого аналитического сигнала от концентрации определяемого компонента. В косвенных методах аналитический сигнал измеряют с целью нахождения конечной точки титрования определяемого компонента подходящим титрантом, то есть используют зависимость измеряемого параметра от объѐматитранта.
Электрохимические методы анализа основаны на изучении и использовании процессов, протекающих на поверхности электрода или в приэлектродном пространстве. Любой электрический параметр (потенциал, электрический ток, количество электричества и др.), функционально связанный с концентрацией определяемого компонента и поддающийся правильному измерению, может служить аналитическим сигналом.
По природе измеряемого аналитического сигнала электрохимические методы анализа разделяют на потенциометрию, вольтамперометрию, кулонометрию и ряд других методов:
Характеристическая зависимость электрохимического сигнала от независимой переменной
| Метод | Измеряемый сигнал | Зависимость сигнала от независимой переменной |
| Потенциометрия, ионометрия | потенциал E = f(C) С-концентрация анализируемого вещества | |
| Потенциометрическое титрование | потенциал E = f(V), V- объем реагента-титранта |
 |
| полярография, вольтамперометрия | ток I = f(E), E – потенциал поляризации электрода |  |
| инверсионная вольтамперометрия | ток I n = f(E) |  |
| хронопотенциометрия | потенциал E =f(t), t – время поляризации электрода при I=const. |
 |
| амперометрическое титрование с одним индикаторным электродом | ток I = f(V), V – объем реагента-титранта |  |
| амперометрическое титрование с двумя индикаторными электродами | ток I = f(V) V – объем реагента-титранта | |
| кулонометрия | Q = f(C), С – количество вещества |  |
| кондуктометрия | G = f(C), С – концентрация ионов в растворе |  |
| кондуктометрическое титрование | электропроводность G = f(V), V – объем реагента-титранта |  |
Потенциометрия
В основе потенциометрических измерений лежит зависимость равновесного потенциала электрода от активности (концентрации) определяемого иона. Для измерений необходимо составить гальванический элемент из подходящего индикаторного электрода и электрода сравнения, а также иметь прибор для измерения потенциала индикаторного электрода (ЭДС гальванического элемента), в условиях близких к термодинамическим, когда индикаторный электрод имеет равновесный (или близкий к нему) потенциал, то есть без отвода заметного тока от гальванического элемента при замыкании цепи. При этом нельзя использовать обычный вольтметр, а следует применять потенциометр - электронный прибор с большим входным сопротивлением (1011 - 1012 Ом), что исключает протекание электродных электрохимических реакций и возникновение тока в цепи.
Индикаторный электрод – это электрод, потенциал которого зависит от активности (концентрации) определяемого иона в анализируемом растворе.
Электрод сравнения – это электрод, потенциал которого в условиях проведения анализа остается постоянным. По отношению к электроду сравнения измеряют потенциал индикаторного электродаЕ (ЭДС гальванического элемента).
В потенциометрии используют два основных класса индикаторных электродов – электронообменные и ионообменные.
Электронообменныеэлетроды – это электроды, на поверхности которых протекают электродные реакции с участием электронов. К таким электродам относятся электроды первого и второго рода, окислительно-восстановительные электроды.
Электроды первого рода – это электроды, обратимые по катиону, общему с материалом электрода, например, металл М, погруженный в раствор соли того же металла. На поверхности такого электрода протекает обратимая реакция M n+ + ne ↔ M и его реальный потенциал зависит от активности (концентрации) катионов металла в растворе в соответствии с уравнением Нернста:

Для температуры 250С (298 K) и для условий, когда активность ионов приблизительно равна концентрации (γ → 1):

Электроды первого рода могут быть изготовлены из различных металлов, например, Ag (серебряный), Cu (медный), Zn (цинковый), Pb (свинцовый) и др.
Схематически электроды первого рода записывают как М | M n + , где вертикальной линией показана граница твердой (электрод) и жидкой (раствор) фаз. Например, серебряный электрод, погруженный в раствор нитрата серебра изображают следующим образом – Ag | Ag+; при необходимости указывают концентрацию электролита – Ag | AgNO 3 (0,1 M).
К электродам первого рода относится и газовый водородный электрод Pt(H 2) | H+ (2Н + + 2е ↔ Н 2 , Е 0 = 0):

Электроды второго рода – это электроды, обратимые по аниону, например, металл, покрытый малорастворимой солью этого металла, погруженный в раствор, содержащий анион этой малорастворимой соли M, MA | А n- . На поверхности такого электрода протекает обратимая реакция MА + ne ↔ M + А n- и его реальный потенциал зависит от активности (концентрации) аниона в растворе в соответствии с уравнением Нернста (приТ = 298 K и γ → 1):

Примерами электродов второго рода служат хлорсеребряный (AgCl + e ↔ Ag + Cl -) и каломельный (Hg 2 Cl 2 + 2e ↔ 2Hg + 2Cl -) электроды:

Окислительно-восстановительные электроды – это электроды, которые состоят из инертного материала (платина, золото, графит, стеклоуглерод и др.), погруженного в раствор, содержащий окисленную (Ок) и восстановленную (Вос) формы определяемого вещества. На поверхности такого электрода протекает обратимая реакция Ок + ne ↔ Вос и его реальный потенциал зависит от активности (концентрации) окисленной и восстановленной форм вещества в растворе в соответствии с уравнением Нернста (приТ = 298 K и γ → 1):

Если в электродной реакции участвуют ионы водорода, то их активность (концентрацию) учитывают в соответствующих уравнениях Нернста для каждого конкретного случая.
Ионообменные электроды – это электроды, на поверхности которых протекают ионообменные реакции. Такие электроды называют также ионселективными или мембранными. Важнейшей составной частью таких электродов является полупроницаемая мембрана – тонкая твердая или жидкая пленка, отделяющая внутреннюю часть электрода (внутренний раствор) от анализируемого и обладающая способностью пропускать только ионы одного вида Х (катионы или анионы). Конструктивно мембранный электрод состоит из внутреннего электрода сравнения (обычно хлорсеребряный) и внутреннего раствора электролита с постоянной концентрацией потенциалопределяющего иона, отделенных от внешнего (исследуемого) раствора чувствительной мембраной.
Реальный потенциал ионселективных электродов, измеренный относительно какого-либо электрода сравнения, зависит от активности тех ионов в растворе, которые сорбируются мембраной:

где const – константа, зависящая от природы мембраны (потенциал асимметрии ) и разности потенциалов внешнего и внутреннего электродов сравнения, n иа (Хn ±) – заряд и активность потенциалопределяющего иона. Если потенциал ионселективного электрода измерен относительно стандартного водородного электрода, то константа является стандартным электродным потенциалом Е 0.
Для мембранных электродов значение крутизны электродной функции может отличаться от теоретической нернстовской величины (0,059 В); в этом случае реальное значение электродной функции θ определяют как тангенс угла наклона градуировочного графика. Тогда:

Потенциал мембранного электрода в растворе, содержащем кроме определяемого иона Х посторонний ион В, влияющий на потенциал электрода, описывается уравнением Никольского (модифицированное уравнение Нернста):

где z – заряд постороннего (мешающего) иона, K Х/В – коэффициент селективности мембранного электрода.
Коэффициент селективности K Х/В характеризует чувствительность мембраны электрода к определяемым ионам Х в присутствии мешающих ионов В. Если K Х/В <1, то электрод селективен относительно ионов Х и, чем меньше числовое значение коэффициента селективности, тем выше селективность электрода по отношению к определяемым ионам и меньше мешающее действие посторонних ионов. Если коэффициент селективности равен 0,01, то это означает, что мешающий ион В оказывает на величину электродного потенциала в 100 раз меньшее влияние, чем определяемый ион той же молярной концентрации.
Рассчитывают коэффициент селективности как отношение активностей (концентраций) определяемого и мешающего ионов, при которых электрод приобретает одинаковый потенциал в растворах этих веществ, с учѐтом их зарядов:

Зная значение коэффициента селективности можно рассчитать концентрацию мешающего иона, влияющую на потенциал ионселективного электрода (пример).
Пример. Какую концентрацию нитратных ионов нужно создать в 1∙10-3 М растворе фторида натрия, чтобы ионселективный фторидный электрод был одинаково чувствителен к обоим ионам, если его коэффициент селективности электрода?

Решение.
Так как, то
Это означает, что концентрация нитратных ионов в анализируемом растворе свыше 0,5 моль/л оказывает значительное влияние на определение фторид-иона в его миллимо-лярных растворах.
Классическим примером ионселективного электрода с твердой мембраной является стеклянный электрод с водородной функцией, служащий для измерения концентрации ионов водорода в растворе (стеклянный рН-электрод). Для таких электродов мембраной служит специальное стекло определѐнного состава, а внутренним электролитом – 0,1 М раствор хлороводородной кислоты:
Ag, AgCl | 0,1 M HCl | стеклянная мембрана | исследуемый раствор
На поверхности стеклянной мембраны происходит ионообменный процесс:
SiO-Na+ (стекло) + Н+ (раствор) → -SiO-H+ (стекло) + Na+ (раствор)
в результате чего устанавливается динамическое равновесие между ионами водорода в стекле и растворе Н+ (стекло) ↔ Н+ (раствор), что приводит к возникновению потенциала:
E = const + θ lga (H+) = const – θ pH
Стеклянный электрод с повышенным содержанием в мембране Al2O3 измеряет ак-тивность ионов натрия в растворе (стеклянный Na-электрод, натрийселективныйэлек-трод). В этом случае внутренним раствором служит 0,1 М раствор хлорида натрия:
Ag, AgCl | 0,1 M NaCl | стеклянная мембрана | исследуемый раствор
На поверхности стеклянной мембраны натрийселективного электрода устанавливается равновесие между ионами натрия в стекле и растворе Na+ (стекло) ↔ Na+ (раствор), что приводит к возникновению потенциала:
E = const + θ lga (Na+) = const – θ pNa
Наиболее совершенным электродом с кристаллической мембраной является фторидселективный электрод, мембрана которого выполнена из пластинки монокристалла фторида лантана (LaF3), активированного для увеличения проводимости фторидом европия (EuF 2):
Ag, AgCl | 0,1 M NaCl, 0,1 M NaF | LaF 3 (EuF 2) | исследуемый раствор
Потенциал фторидного электрода определяется ионообменным процессом на его поверхности F- (мембрана) ↔ F- (раствор):
E = const – θ lga (F-) = const + θ pF
Значения константы и крутизны электродной функции θ для ионселективных электродов определяют из градуировочного графикаЕ ÷ рХ как отрезок на оси ординат и тангенс угла наклона прямой соответственно. Для стеклянного рН-электрода эта операция заменяется настройкой приборов (рН-метров) по стандартным буферным растворам с точно известными значениями рН.
Схематический вид стеклянного и фторидселективного электродов приведены на рисунках:
В паре с индикаторным электродом для измерения его потенциала (ЭДС гальванической ячейки) используют электрод сравнения с известным и устойчивым потенциалом, не зависящим от состава исследуемого раствора. Наиболее часто в качестве электрода сравнения применяют хлорсеребряный и каломельный электроды. Оба электрода относятся к электродам второго рода и характеризуются высокой стабильностью в работе.
Потенциалы хлорсеребряного и каломельного электродов зависят от активности (концентрации) хлорид-ионов (приТ = 298 K и γ → 1):

В качестве электродов сравнения чаще всего применяют электроды с насыщенным раствором хлорида калия – при 250С потенциал насыщенного хлорсеребряного электрода сравнения равен +0,201 В, а насыщенного каломельного +0,247 В (относительно стандартного водородного электрода). Потенциалы для хлорсеребряных и каломельных электродов сравнения, содержащих 1 М и 0,1 М растворы хлорида калия, можно найти в справочных таблицах.
Схематический вид насыщенных хлорсеребряного и каломельного электродов срав-нения приведены на рисунке:

Электроды сравнения хлорсеребряный (а) и каломельный (б)
1 - асбестовое волокно, обеспечивающее контакт с анализируемым раствором
2 - раствор KCl (насыщенный)
3 - отверстие для контакта
4 - раствор KCl (насыщенный), AgCl (тв.)
5 - отверстие для ввода раствора KCl
6 - паста из смеси Hg2Cl2, Hg и КС1 (насыщенный)
Потенциометрический анализ широко применяют для непосредственного определения активности (концентрации) ионов в растворе путем измерения равновесного потенциала индикаторного электрода (ЭДС гальванического элемента) – прямая потенциометрия (ионометрия) , а также для индикации конечной точки титрования (ктт ) по изменению потенциала индикаторного электрода в процессе титрования (потенциометрическое титрование).
Во всех приемахпрямой потенциометрии используется зависимость индикаторного электрода от активности (концентрации) определяемого иона, которая описывается уравнением Нернста. Результаты анализа подразумевают определение концентрации вещества, а не его активности, что возможно при значении коэффициентов активности ионов равных единице (γ → 1) или их постоянном значении (постоянной ионной силе раствора), поэтому в дальнейших рассуждениях используются только концентрационные зависимости.
Концентрация определяемого иона может быть рассчитана по экспериментально найденному потенциалу индикаторного электрода, если для электрода известны постоянная составляющая (стандартный потенциал Е 0) и крутизна электродной функции θ . В этом случае составляется гальванический элемент, состоящий из индикаторного электрода и электрода сравнения, измеряется его ЭДС, рассчитывается потенциал индикаторного электрода (относительно СВЭ) и концентрация определяемого иона.
В методеградуировочного графика готовят серию стандартных растворов с известной концентрацией определяемого иона и постоянной ионной силой, измеряют потенциал индикаторного электрода относительно электрода сравнения (ЭДС гальванического элемента) в этих растворах и по полученным данным строят зависимость Е ÷ рС (А) (градуировочный график). Затем измеряют потенциал индикаторного электрода в анализируемом растворе Е х (в тех же условиях) и по графику определяют рС х(А) и рассчитывают концентрацию определяемого вещества в анализируемом растворе.
В методе стандарта (сравнения) измеряют потенциал индикаторного электрода в анализируемом растворе (Е х) и в стандартном растворе определяемого вещества (Е ст). Расчет концентрации определяемого иона проводят исходя из уравнений Нернста для анализируемой пробы и стандартного образца. Крутизна электродной функции для индикаторного электрода θ
При использовании метода добавок сначала измеряют потенциал индикаторного электрода в анализируемом растворе (Е х), затем добавляют к нему определенный объём стандартного раствора определяемого вещества и измеряют потенциал электрода в полученном растворе с добавкой (Е х+д). Расчет концентрации определяемого иона проводят исходя из уравнений Нернста для анализируемой пробы и пробы с добавкой. Крутизна электродной функции для индикаторного электрода θ должна быть известна или определена заранее по градуировочному графику.
При потенциометрическом титровании измеряют и записывают ЭДС электрохимической ячейки (потенциал индикаторного электрода) после добавления каждой порции титранта. Затем по полученным результатам строят кривые титрования – интегральную в координатах E ÷ V(а) и дифференциальную в координатах ∆E /∆V ÷ V (б) , и определяют конечную точку титрования (ктт) графическим способом:

В потенциометрическом титровании используют все основные типы химических реакций – кислотно-основные, окислительно-восстановительные, осаждения и комплексообразования. К ним предъявляются те же требования, что и в визуальной титриметрии, дополненные наличием подходящего индикаторного электрода для фиксации изменения концентрации потенциалопределяющих ионов в ходе титрования.
Погрешность определения при проведении потенциометрического титрования составляет 0,5-1%, что существенно ниже, чем при прямых потенциометрических измерениях (2-10%), однако, при этом наблюдаются более высокие пределы обнаружения – больше 10 -4 моль/л.
Кулонометрия
Кулонометрия объединяет методы анализа, основанные на измерении количества электричества, затраченного на электрохимическую реакцию. Электрохимическая реакция приводит к количественному электропревращению (окислению или восстановлению) определяемого вещества на рабочем электроде (прямая кулонометрия) или к получению промежуточного реагента (титранта), который стехиометрически реагирует с определяемым веществом (косвенная кулонометрия, кулонометрическое титрование).
В основе кулонометрических методов лежит закон Фарадея , который устанавливает связь между количеством электропревращенного (окисленного или восстановленного) вещества и количеством израсходованного при этом электричества:

где m – масса электропревращенного вещества,г; Q – количество электричества, затраченного на электропревращение вещества, Кл; F – число Фарадея, равное количеству электричества, необходимого для электропревращения одного моль-эквивалента вещества, 96500 Кл/моль; М – молярная масса вещества, г/моль; n – число электронов, участвующих в электрохимической реакции.
Необходимым условием проведения кулонометрического анализа является практически полное расходование электричества на превращение определяемого вещества, то есть электрохимическая реакция должна протекать без побочных процессов со 100% вы-ходом по току.
На практике кулонометрический анализ реализуется в двух вариантах – при постоянном потенциале (потенциостатическаякулонометрия ) и при постоянной силе тока(амперостатическаякулонометрия ).
Потенциостатическуюкулонометрию применяют для прямых кулонометрических измерений, когда электролизу подвергается непосредственно определяемое вещество. При этом потенциал рабочего электрода с помощью потенциостатов поддерживается постоянным и его значение выбирают на основе поляризационных кривых в области предельного тока определяемого вещества. В процессе электролиза при постоянном потенциале сила тока уменьшается в соответствии с уменьшением концентрации электроактивного вещества по экспоненциальному закону:

где Ι – сила тока в момент времени t , А; Ι 0 – сила тока в начальный момент электролиза, А; k – константа, зависящая от условий электролиза.
Электролиз ведут до достижения остаточного тока Ι , величина которого определяется требуемой точностью – для допустимой погрешности 0,1% электролиз можно считать завершенным при Ι = 0,001Ι 0 . Для сокращения времени электролиза следует применять рабочий электрод большой поверхности при интенсивном перемешивании анализируемого раствора.
Общее количество электричества Q , необходимое для электропревращения определяемого вещества, определяется уравнением:

Определить количество электричества можно измерением площади под кривой «ток – время» с помощью механических или электронных интеграторов, либо с помощью химических кулонометров. Кулонометр – это электролитическая ячейка, в которой со 100% выходом по току протекает электрохимическая реакция известной стехиометрии. Кулонометр включают последовательно с исследуемой кулонометрической ячейкой, поэтому за время электролиза через обе ячейки протекает одинаковое количество электричества. Если по окончании электролиза измерить количество (массу) образовавшегося в кулонометре вещества, то по закону Фарадея можно рассчитать количество электричества. Чаще всего применяют серебряный, медный и газовые кулонометры.
Амперостатическую кулонометрию применяют для кулонометрического титрования при постоянном токе, в процессе которого определяемое вещество реагирует с титрантом, образующимся в результате электрохимической реакции на рабочем электроде, а потому, называемый электрогенерированным титрантом .
Для обеспечения 100%-ного выхода по току необходим значительный избыток вспомогательного вещества, из которого генерируется титрант, что исключает протекание конкурирующих реакций на рабочем электроде. При этом титрант генерируется в количестве, эквивалентном определяемому веществу, и по количеству электричества, затраченного на генерацию титранта, можно рассчитать содержание определяемого вещества.
Количество электричества Q в кулонометрии при постоянной силе тока Ι рассчитывают по формуле:

где t – время электролиза, для определения которого пригодны практически все способы установления конечной точки в титриметрии (визуальные – индикаторы, инструментальные – потенциометрия, амперометрия, фотометрия). При силе тока в амперах и времени электролиза в секундах получаем количество электричества в кулонах (пример).
Пример. На кулонометрическое титрование раствора аскорбиновой кислоты иодом, генерируемым из иодида калия током силой 5,00 мА, потребовалось 8 мин 40 с. Рассчитать массу аскорбиновой кислоты в анализируемом растворе. Предложить способ фиксирования конечной точки титрования.
Решение. Количество электричества, затраченное на окисление иодида и, соответственно, аскорбиновой кислоты равно:
Q = Ι·t = 5,00∙10 -3 ∙520 = 2,60 Кл.
Аскорбиновая кислота окисляется иодом до дегидроаскорбиновой кислоты с отдачей двух электронов (С 6 Н 8 О 6 – 2е → С 6 Н 6 О 6 + 2Н +), тогда по закону Фарадея:

Конечная точка титрования определяется по появлению избытка иода в растворе. Следовательно, фиксировать ее можно визуально с помощью крахмала, добавленного в анализируемый раствор (появление синей окраски), амперометрически с ртутным капающим или платиновым микроэлектродом по появлению предельного тока иода, потенциометрически по резкому увеличению потенциала платинового электрода.
Вольтамперометрия
Вольтамперометрический метод анализа основан на использовании явления поляризации микроэлектрода, получении и интерпретации вольтамперных (поляризационных) кривых, отражающих зависимость силы тока от приложенного напряжения. Вольтамперная кривая (вольтамперограмма) позволяет одновременно получить качественную и количественную информацию о веществах, восстанавливающихся или окисляющихся на микроэлектроде (деполяризаторах), а также о характере электродного процесса. Современная вольтамперометрия – высокочувствительный и экспрессный метод определения веществ, пригодный для анализа различных объектов неорганической и органической природы, в том числе и фармацевтических препаратов. Минимально определяемая концентрация в вольтамперометрии достигает значений 10 -8 моль/л при погрешности метода менее 5%. Вольтамперометрия при оптимальных условиях эксперимента позволяет в анализируемом растворе определять несколько компонентов одновременно.
В вольтамперометрии используют два электрода – рабочий поляризуемый электрод с малой поверхностью (индикаторный микроэлектрод) и вспомогательный неполяризуемый электрод с большой поверхностью (электрод сравнения). Рабочими электродами служат микроэлектроды из ртути (ртутный капающий электрод, РКЭ), платины (ПЭ) и токопроводящих углеродных материалов (графит, стеклоуглерод).
При прохождении постоянного тока через электролитическую ячейку процесс характеризуется соотношением (закон Ома для раствора электролита):
Е = Ea – Eк + IR
Где Е – приложенное внешнее напряжение; Еа – потенциал анода; Ек – потенциал катода; I – ток в цепи; R – внутреннее сопротивление электролитической ячейки.
При вольтамперометрических измерениях анализируемый раствор содержит индифферентный (фоновый) электролит большой концентрации (в 100 раз и более превышающей концентрацию определяемого вещества – сопротивление раствора мало), а ток в вольтамперометрии не превышает 10 -5 А, поэтому падением напряжения в ячейке IR можно пренебречь.
Поскольку в вольтамперометрии один из электродов (вспомогательный) не поляризуется и для него потенциал остается постоянным (его можно принять равным нулю), подаваемое на ячейку напряжение проявляется в изменении потенциала только рабочего электрода и тогда Е = Ea для рабочего микроанода (анодная поляризация ) и Е = -Eк для рабочего микрокатода (катодная поляризация ). Таким образом, регистрируемая вольтамперная кривая отражает электрохимический процесс, происходящий только на рабочем электроде. Если в растворе присутствуют вещества, способные электрохимически восстанавливаться или окислятся, то при наложении на ячейку линейно изменяющегося напряжения вольтамперограмма имеет форму волны 1 (в отсутствии электрохимической реакции зависимость тока от напряжения линейна 2 в соответствии с законом Ома):

Раздел вольтамперометрии, в котором рабочим микроэлектродом служит РКЭ называют полярографией , в честь чешского электрохимика Я.Гейровского, предложившего этот метод в 1922 году. Вольтамперограммы, полученные в ячейке с ртутным капающим электродом, называют полярограммами.
Для регистрации классических полярограмм ячейку с РКЭ (рабочий электрод) и насыщенным каломельным электродом (вспомогательный электрод, электрод сравнения) присоединяют к источнику постоянного напряжения и изменяют потенциал со скоростью 2-5 мВ/с.
Ртутный капающий электрод является практически идеально поляризуемым в широком диапазоне потенциалов, ограниченном в анодной области электродными реакциями окисления ртути (+0,4 В), а в катодной реакциями восстановления ионов водорода (от -1 до -1,5 Вв зависимости от кислотности среды) или катионов фона (от -2 В для катионов щелочных металлов до -2,5 В для R 4 N +). Это позволяет изучать и определять на РКЭ вещества, восстанавливающиеся при очень высоких отрицательных потенциалах, что невозможно на электродах из других материалов. Следует отметить, что здесь и далее значения потенциалов приведены относительно насыщенного каломельного электрода и при необходимости могут быть пересчитаны по отношению к другому электроду сравнения, например, насыщенному хлорсеребряному.
Перед регистрацией полярограммы на РКЭ необходимо удалить растворенный кислород, поскольку он электроактивен в отрицательной области потенциалов, давая две волны восстановления при -0,2 и -0,9 В. Сделать это можно, насыщая раствор инертным газом (азот, аргон, гелий). Из щелочных растворов кислород удаляют с помощью сульфита натрия (O 2 + 2Na 2 SO 3 → 2Na 2 SO 4).
Классическая полярограмма (полярографическая волна) в идеализированном виде представлена ниже:

Основными характеристиками полярографической волны являются величина диффузионного тока (I д, мкА), потенциал полуволны (Е 1/2 , В) – потенциал, при котором ток равен половине диффузионного, и наклон восходящего участка (0,059/n – крутизна электродной функции). Эти параметры позволяют использовать полярографию как метод анализа (сила тока пропорциональна концентрации) и исследования (потенциал полуволны и электродная функция зависят от природы вещества).
На начальном участке полярографической волны (А-Б) ток с изменением потенциала возрастает очень медленно – это так называемый остаточный ток (I ост). Основной вклад в остаточный ток вносит формирование двойного электрического слоя (ток заряжения ), который невозможно исключить и величина которого возрастает с увеличением потенциала. Вторым слагаемым остаточного тока является ток, обусловленный электроактивными примесями, который можно уменьшить применяя чистые реактивы и воду.
При достижении точки Б (потенциал выделения – при восстановлении на катоде потенциал выделения называют потенциалом восстановления Е вос, при окислении на аноде – потенциалом окисления Е ок) на электроде начинается электрохимическая реакция, в которую вступает электроактивное вещество (деполяризатор), в результате чего ток резко возрастает (участок Б-В) до некоторого предельного значения, оставаясь затем практически постоянным (участок В-Г). Ток, соответствующий этому участку называют предельным током (I пр), а разность между предельным и остаточным током составляет диффузионный ток (I д = I пр – I ост). На участке В-Г при увеличении потенциала предельный и остаточный токи незначительно возрастают, а значение диффузионного тока остается постоянным. Подъем тока в точке Г обусловлен новой электрохимической реакцией (например, восстановлением катионов фонового электролита).
Диффузионный ток получил свое название вследствие того, что в данной области потенциалов в результате электрохимической реакции в приэлектродном слое наблюдается практически полное отсутствие деполяризатора и его обогащение веществом происходит за счет диффузии деполяризатора из глубины раствора, где его концентрация остается постоянной. Поскольку скорость диффузии в данных конкретных условиях остается постоянной, то и диффузионный ток сохраняет постоянство своего значения.
Зависимость величины диффузионного тока от концентрации деполяризатора для р.к.э. выражается уравнением Ильковича:
I d = 605nD 1/2 m 2/3 t 1/6 c
где D – коэффициент диффузии электроактивного иона; n – число электронов, участвующих в реакции; m 2/3 t 1/6 – характеристика капилляра, из которого вытекает ртуть; с - концентрация определяемого вещества (деполяризатора).
При работе с одним и тем же капилляром и деполяризатором значение 605nD 1/2 m 2/3 t 1/6 = const, поэтому между высотой волны и концентрацией вещества имеется линейная зависимость
На этой линейной зависимости основан количественный полярографический анализ. Взаимосвязь между потенциалом электрода и возникающим током описывается уравнением полярографической волны (уравнение Ильковича-Гейровского):

где Е и I – соответственно потенциал и величина тока для данной точки полярографической кривой; I d - величина диффузионного тока; Е 1/2 – потенциал полуволны.
Е 1/2 - это потенциал, при котором достигается величина тока, равная половине I d . Он не зависит от концентрации деполяризатора. Е 1/2 очень близки к нормальному редокс-потенциалу системы (Ео), то есть является качественной характеристикой, определяющейся только природой восстанавливающихся ионов и по которым можно установить качественный состав анализируемого раствора.
Полярограмма (вольтамперограмма) содержит ценную аналитическую информацию – потенциал полуволны Е 1/2 является качественной характеристикой деполяризатора (качественный аналитический сигнал), в то время как диффузионный ток I д линейно связан с концентрацией определяемого вещества в объёме анализируемого раствора (количественный аналитический сигнал) – I д = KС .
Величина Е 1/2 может быть рассчитана из уравнения полярографической волны или определена графически:

Найденное значение Е 1/2 с учетом использованного фонового электролита позволяет на основании табличных данных идентифицировать деполяризатор. Если в анализируемом растворе находится несколько веществ, потенциалы полуволн которых различаются более чем на 0,2 В, то на полярограмме будет не одна волна, а несколько – по числу электроактивных частиц. При этом следует иметь в виду, что восстановление (окисление) многозарядных частиц может происходить ступенчато, давая несколько волн.
Для исключения перемещения вещества к электроду за счет тепловой и механической конвекции (перемешивания) измерение осуществляется в термостатированном растворе и в отсутствии перемешивания. Устранению электростатического притяжения деполяризатора полем электрода (миграции) способствует большой избыток электронеактивного фонового электролита, ионы которого экранируют заряд электрода, уменьшая движущую силу миграции практически до нуля.
При использовании ртутного капающего электрода на полярограмме наблюдаются осцилляции тока (его периодическое небольшое увеличение и уменьшение). Каждая такая осцилляция соответствует возникновению, росту и отрыву капли ртути от капилляра микроэлектрода. В полярографах предусмотрены устройства для устранения осцилляций.
Полярограммы могут быть искажены за счет полярографических максимумов – резкого возрастания тока выше его предельного значения с последующим спадом:

Появление максимумов обусловлено перемешиванием раствора в результате движения поверхности капли ртути из-за неравномерного распределения заряда, а, соответственно, и поверхностного натяжения (максимумы I рода), а также появлений завихрений при вытекании ртути из капилляра (максимумы II рода). Максимумы искажают полярограмму и затрудняют еѐ расшифровку. Для удаления максимумов I рода вводят поверхностно-активное вещество (например, агар-агар, желатин, камфару, фуксин, синтетические ПАВ), которое, адсорбируясь на поверхности ртутной капли, выравнивает поверхностное натяжение и устраняет движение поверхностных слоѐв ртути. Для удаления максимумов II рода достаточно уменьшить давление ртути в капилляре, снизив высоту ртутного столба.
Вольтамперометрия с твердыми рабочими электродами отличается от полярографии с использованием РКЭ другим диапазоном поляризации микроэлектрода. Как было показано выше, ртутный капающий электрод вследствие высокого перенапряжения водорода на нём можно использовать в области высоких отрицательных потенциалов, но из-за анодного растворения ртути при +0,4 В он не может быть применѐн для исследований в области положительных потенциалов. На графите и платине разряд ионов водорода протекает значительно легче, поэтому область их поляризации ограничена значительно более низкими отрицательными потенциалами (-0,4 и -0,1 В соответственно). В то же время в области анодных потенциалов платиновый и графитовый электроды пригодны до потенциала +1,4 В (далее начинается электрохимическая реакция окисления кислорода воды 2Н 2 О – 4е → О 2 + 4Н +), что делает их пригодными для исследований в диапазоне положительных потенциалов.
В отличие от РКЭ во время регистрации вольтамперограммы поверхность твердого микроэлектрода не возобновляется и легко загрязняется продуктами электродной реакции, что приводит к понижению воспроизводимости и точности результатов, поэтому перед регистрацией каждой вольтамперограммы следует проводить очистку поверхности микроэлектрода.
Стационарные твердые электроды не нашли широкого применения в вольтамперометрии из-за медленного установления предельного тока, что приводит к искажению формы вольтамперограммы, однако, на вращающихся микроэлектродах в приэлектродном слое возникают условия для стационарной диффузии, поэтому сила тока устанавливается быстро и вольтамперограмма имеет ту же форму, что и в случае РКЭ.
Величина предельного диффузионного тока на вращающемся дисковом электроде (не зависимо от материала) описывается уравнением конвективной диффузии (Левича):
I d = 0.62nFSD 2/3 w 1/2 n -1/6 c
где n - число электронов, участвующих в электродном процессе;
F – число Фарадея (96500 кулонов);
S - площадь электрода;
D – коэффициент диффузии деполяризатора;
w - угловая скорость вращения электрода;
n - кинематическая вязкость исследуемого раствора;
с - концентрация деполяризатора, моль/л.
При затруднениях в расшифровке полярограмм применяют метод «свидетеля» – после регистрации полярограммы анализируемого раствора, к нему в электролитическую ячейку поочередно добавляют стандартные растворы предполагаемых соединений. Если предположение было верным, то увеличивается высота волны соответствующего вещества, при неверном предположении появится дополнительная волна при другом потенциале.
Определить концентрацию деполяризатора в анализируемом растворе можно методами градуировочного графика, методом стандарта (сравнения) и методом добавок. При этом во всех случаях следует использовать стандартные растворы, состав которых максимально приближен к составу анализируемого раствора, а условия регистрации полярограмм должны быть одинаковы. Методы применимы в интервале концентраций, где строго соблюдается прямо пропорциональная зависимость диффузионного тока от концентрации деполяризатора. На практике при количественных определениях, как правило, не фиксируют величину диффузионного тока в мкА, а измеряют высоту полярографической волны h , как указано на предыдущем рисунке, которая также является линейной функцией от концентрации h = KC.
По методу градуировочного графика регистрируют полярограммы серии стандартных растворов и строят градуировочный график в координатах h ÷ C (или I д ÷ С ), по которому для найденного значения h x в анализируемом растворе находят концентрацию определяемого вещества в нѐм С х.
В методе стандарта (сравнения) в одних и тех же условиях записывают полярограммы анализируемого и стандартного растворов определяемого вещества с концентрациями С х и С ст, тогда:

При использовании метода добавок сначала записывают полярограмму анализируемого раствора объемомV x с концентрацией С х и измеряют высоту волны h x. Затем в электролитическую ячейку к анализируемому раствору добавляют определенный объѐм стандартного раствора определяемого вещества V д с концентрацией С д (предпочтительно, чтобы V x>>V д и С х<С д), записывают полярограмму раствора с концентрацией С х+д и из-меряют высоту полученной волны h х+д. Несложные преобразования позволяют по этим данным позволяют рассчитать концентрацию определяемого вещества в анализируемом растворе (пример).
Пример. При полярографировании 10,0 мл раствора никотинамида получена волна высотой 38 мм. После добавления к этому раствору 1,50 мл стандартного раствора, содержащего 2,00 мг/мл никотинамида, волна увеличилась до 80,5 мм. Рассчитать содержание препарата (мг/мл) в анализируемом растворе.
Решение. Высота волны никотинамида в анализируемом растворе h x в соответствии с уравнением Ильковича равна:

а после добавки стандартного раствора (h х+д):

Если первое уравнение почленно разделить на второе, то получим:

Решая уравнение относительно С х и подставив значения величин из условия задачи.
«Электрохимические методы анализа и их современное аппаратурное оформление: обзор WEB–сайтов фирм–продавцов химико-аналитического оборудования»
Введение
Глава 1. Классификация электрохимических методов
1.1 Вольтамперометрия
1.2 Кондуктометрия
1.3 Потенциометрия
1.4 Амперометрия
1.5 Кулонометрия
1.6 Другие электрохимические явления и методы
1.7 Прикладная электрохимия
Глава 2. Электрохимические методы анализа и их роль в охране окружающей среды
Глава 3. Приборы на основе электрохимических методов анализа
Глава 4. Обзор WEB – сайтов фирм – продавцов химико-аналитического оборудования
Литература
ВВЕДЕНИЕ
Электрохимические методы анализа
(электроанализ), в основе которых лежат электрохимические процессы, занимают
достойное место среди методов контроля состояния окружающей среды, так как
способны обеспечить определение огромного числа как неорганических, так и
органических экологически опасных веществ. Для них характерны высокая
чувствительность и селективность, быстрота отклика на изменение состава
анализируемого объекта, легкость автоматизации и возможность дистанционного
управления. И, наконец, они не требуют дорогостоящего аналитического
оборудования и могут применяться в лабораторных, производственных и полевых
условиях. Непосредственное отношение к рассматриваемой проблеме имеют три
электроаналитических метода: вольтамперометрия, кулонометрия и потенциометрия.
ГЛАВА 1. КЛАССИФИКАЦИЯ ЭЛЕКТРОХИМИЧЕСКИХ
МЕТОДОВ
Электрохимические методы анализа (ЭМА) основаны на исследовании процессов, протекающих на поверхности электрода или в приэлектродном пространстве. Аналитическим сигналом служит электрический параметр (потенциал, сила тока, сопротивление и др.), функционально связанный с концентрацией определяемого компонента раствора и поддающийся правильному измерению.
Классификация ЭМА, предлагаемая ИЮПАК, за последние десятилетия претерпела определенные изменения, в нее внесены уточнения (пояснения) и дополнения.
Существенное внимание уделяется электрохимическим ячейкам и датчикам аналитического сигнала (электродным системам, различным электрохимическим сенсорам), именно эти первичные электрохимические преобразователи определяют аналитические возможности любого метода. В настоящее время не представляет проблемы самая совершенная и быстрая обработка сигнала от датчика, расчет статистических характеристик как исходного сигнала, так и результатов всего анализа в целом. Именно поэтому важно получить достоверный исходный сигнал, чтобы прокалибровать его в единицах концентрации.
Согласно общей классификации, предложенной
ИЮПАК, ЭМА подразделяются на методы, в которых возбуждаемый электрический сигнал постоянен или равен нулю и на методы, в которых возбуждаемый сигнал меняется во времени. Эти методы классифицируются следующим образом:
вольтамперометрические – voltammetry, I ≠ 0; E = f(t) ;
потенциометрические – potentiometry, (I = 0);
амперометрические – amperometry (I ≠ 0; E = const);
хронопотенциометрические, E = f(t) ; I = const;
импедансные, или кондуктометрические - измерения, использующие наложение переменного напряжения малой амплитуды; другие, комбинированные (например, спектроэлектрохимические).
1.1 ВОЛЬТАМПЕРОМЕТРИЯ
ВОЛЬТАМПЕРОМЕТРИЯ - совокупность электрохимических методов исследования и анализа, основанных на изучении зависимости силы тока в электролитические ячейке от потенциала погруженного в анализируемый раствор индикаторного микроэлектрода, на котором реагирует исследуемое электрохимически активное (электроактивное) вещество. В ячейку помещают помимо индикаторного вспомогательный электрод со значительно большей поверхностью, чтобы при прохождении тока его потенциал практически не менялся (неполяризующийся электрод). Разность потенциалов индикаторного и вспомогательного электродов Е описывается уравнением Е = U - IR, где U - поляризующее напряжение, R-сопротивление раствора. В анализируемый раствор вводят в большой концентрации индифферентный электролит (фон), чтобы, во-первых, уменьшить величину R и, во-вторых, исключить миграционный ток, вызываемый действием электрического поля на электроактивные вещества (устар. - деполяризаторы). При низких концентрациях этих веществ омическое падение напряжения IR в растворе очень мало. Для полной компенсации омического падения напряжения применяют потенциостатирование и трехэлектродные ячейки, содержащие дополнительно электрод сравнения. В этих условиях
В качестве индикаторных микроэлектродов используют стационарные и вращающиеся - из металла (ртуть, серебро, золото, платина), углеродных материалов (напр., графит), а также капающие электроды (из ртути, амальгам, галлия). Последние представляют собой капилляры, из которых по каплям вытекает жидкий металл. Вольтамперометрия с использованием капающих электродов, потенциал которых меняется медленно и линейно, наз. полярографией (метод предложен Я. Гейровским в 1922). Электродами сравнения служат обычно электроды второго рода, напр. каломельный или хлоросеребряный (см. Электроды сравнения). Кривые зависимости I =f(E) или I =f(U) (вольтамперограммы) регистрируют специальными приборами - полярографами разных конструкций.
Вольтамперограммы, полученные с помощью вращающегося или капающего электрода при монотонном изменении (линейной развертке) напряжения, имеют вид, схематически представленный на рисунке. Участок увеличения тока наз. волной. Волны м. б. анодными, если электроактивное вещество окисляется, или катодными, если оно восстанавливается. Когда в растворе присутствуют окисленная (Ох) и восстановленная (Red) формы вещества, достаточно быстро (обратимо) реагирующие на микроэлектроде, на вольтамперограмме наблюдается непрерывная катодно-анодная волна, пересекающая ось абсцисс при потенциале, соответствующем окислительно-восстановитановительному потенциалу системы Ox/Red в данной среде. Если электрохимическая реакция на микроэлектроде медленная (необратимая), на вольтамперограмме наблюдаются анодная волна окисления восстановленной формы вещества и катодная волна восстановления окисленной формы (при более отрицат. потенциале). Образование площадки предельного тока на вольтамперограмме связано либо с ограниченной скоростью массопереноса электроактивного вещества к поверхности электрода путем конвективной диффузии (предельный диффузионный ток, I d), либо с ограниченной скоростью образования электроактивного вещества из определяемого компонента в растворе. Такой ток называется предельным кинетическим, а его сила пропорциональна концентрации этого компонента.
Форма волны для обратимой электрохимические реакции описывается уравнением:
где R-газовая
постоянная, Т-абсолютная температура, E 1/2 -потенциал полуволны, т.е.
потенциал, соответствующий половине высоты волны (I d /2;). Значение E 1/2
характерно для данного электроактивного вещества и используется для его идентификации.
Когда электрохимические реакции предшествует адсорбция определяемого вещества
на поверхности электрода, на вольтамперограммах наблюдаются не волны, а пики,
что связано с экстремальной зависимостью адсорбции от потенциала электрода. На
вольтамперограммах, зарегистрированных при линейном изменении (развертке)
потенциала со стационарным электродом или на одной капле капающего электрода
(устар. - осциллографич. полярограмме), также наблюдаются пики, нисходящая
ветвь которых определяется обеднением приэлектродного слоя раствора
электроактивным веществом. Высота пика при этом пропорциональна концентрации
электроактивного вещества. В полярографии предельный диффузионный ток (в мкА),
усредненный по времени жизни капли, описывается уравнением Ильковича:
где n-число электронов, участвующих в электрохимической реакции, С-концентрация электроактивного вещества (мМ), D-eгo коэффициент диффузии (см 2 /с),время жизни ртутной капли (с), m-скорость вытекания ртути (мг/с).
С вращающимся
дисковым электродом предельный диффузионный ток рассчитывают по уравнению:
где S-площадь поверхности электрода (см 2),-круговая частота вращения электрода (рад/с), v-кинематическая вязкость раствора (см 2 /с), F-число Фарадея (Кл/моль).
Циклическая вольтамперометрия (вольтамперометрия с относительно быстрой треугольной разверткой потенциала) позволяет изучать кинетику и механизм электродных процессов путем наблюдения на экране осциллографической трубки с послесвечением одновременно вольтамперограмм с анодной и катодной разверткой потенциала, отражающих, в частности, и электрохимические реакции продуктов электролиза.
Нижняя граница определяемых концентраций С н в методах В. с линейной разверткой потенциала составляет 10 -5 -10 -6 М. Для ее снижения до 10-7 -10 -8 М используют усовершенствованные инструментальные варианты - переменно-токовую и дифференциальную импульсную вольтамперометрию.
В первом из этих вариантов на постоянную составляющую напряжения поляризации налагают переменную составляющую небольшой амплитуды синусоидальной, прямоугольной (квадратноволновая вольтамперометрия), трапециевидной или треугольной формы с частотой обычно в интервале 20-225 Гц. Во втором варианте на постоянную составляющую напряжения поляризации налагают импульсы напряжения одинаковой величины (2-100 мВ) длительностью 4-80 мс с частотой, равной частоте капания ртутного капающего электрода, или с частотой 0,3-1,0 Гц при использовании стационарных электродов. В обоих вариантах регистрируют зависимость от U или Е переменной составляющей тока с фазовой или временной селекцией. Вольтамперограммы при этом имеют вид первой производной обычной вольтамперометрической волны. Высота пика на них пропорциональна концентрации электроактивного вещества, а потенциал пика служит для идентификации этого вещества по справочным данным.
Пики различных электроактивных веществ, как правило, лучше разрешаются, чем соответствующие вольтамперометрические волны, причем высота пика в случае необратимой электрохимической реакции в 5-20 раз меньше высоты пика в случае обратимой реакции, что также обусловливает повышенную разрешающую способность этих вариантов вольтамперометрии. Например, необратимо восстанавливающийся кислород практически не мешает определению электроактивных веществ методом переменно-токовой вольтамперометрии. Пики на переменно-токовых вольтамперограммах отражают не только электрохимические реакции электроактивных веществ, но и процессы адсорбции - десорбции неэлектроактивных веществ на поверхности электрода (пики нефарадеевского адмиттанса, устар. - тенсамметрич. пики).
Для всех вариантов вольтамперометрии используют способ снижения С н, основанный на предварительном электрохимическом, адсорбционном или химическом накоплении определяемого компонента раствора на поверхности или в объеме стационарного микроэлектрода, с последующей регистрацией вольтамперограммы, отражающей электрохимическую реакцию продукта накопления. Эту разновидность вольтамперометрии называют инверсионной (устар. название инверсионной В. с накоплением на стационарном ртутном микроэлектроде - амальгамная полярография с накоплением). В инверсионной вольтамперометрии с предварительным накоплением С н достигает 10 -9 -10 -11 М. Минимальные значения С н получают, используя тонкопленочные ртутные индикаторные электроды, в т.ч. ртутно-графитовые, состоящие из мельчайших капелек ртути, электролитически выделенных на подложку из специально обработанного графита.
Для фазового и элементного анализа твердых тел используют инверсионную вольтамперометрию с электроактивными угольными электродами (т. наз. минерально-угольными пастовыми электродами). Их готовят из смеси угольного порошка, исследуемого порошкообразного вещества и инертного связующего, напр. вазелинового масла. Разработан вариант этого метода, который дает возможность проводить анализ и определять толщину металлических покрытий. В этом случае используют специальное устройство (прижимная ячейка), позволяющее регистрировать вольтамперограмму, пользуясь каплей фонового электролита, нанесенного на исследуемую поверхность.
Применение
Вольтамперометрию
применяют: для количественного анализа неорганических и органических веществ в
очень широком интервале содержаний - от 10 -10 % до десятков %; для
исследования кинетики и механизма электродных процессов, включая стадию
переноса электрона, предшествующие и последующие химические реакции, адсорбцию
исходных продуктов и продуктов электрохимических реакций и т. п.; для изучения
строения двойного электрического слоя с, равновесия комплексообразования в
растворе, образования и диссоциации интерметаллических соединений в ртути и на
поверхности твердых электродов; для выбора условий амперометрического
титрования и др.
1.2 Кондуктометрия
Кондуктометрия - основана на измерении электропроводности раствора и применяется для определения концентрации солей, кислот, оснований и т.д. При кондуктометрических определениях обычно используют электроды из одинаковых материалов, а условия их проведения подбирают таким образом, чтобы свести к минимуму вклад скачков потенциала на обеих границах раздела электрод/электролит (например, используют переменный ток высокой частоты). В этом случае основной вклад в измеряемый потенциал ячейки вносит омическое падение напряжения IR, где R – сопротивление раствора. Электропроводность однокомпонентного раствора можно связать с его концентрацией, а измерение электропроводности электролитов сложного состава позволяет оценить общее содержание ионов в растворе и применяется, например, при контроле качества дистиллированной или деионизованной воды. В другой разновидности кондуктометрии – кондуктометрическом титровании – к анализируемому раствору порциями добавляют известный реагент и следят за изменением электропроводности. Точка эквивалентности, в которой отмечается резкое изменение электропроводности, определяется из графика зависимости этой величины от объема добавленного реагента.
1.3 Потенциометрия
Потенциометрия - применяется для определения различных физико-химических параметров исходя из данных о потенциале гальванического элемента. Электродный потенциал в отсутствие тока в электрохимической цепи, измеренный относительно электрода сравнения, связан с концентрацией раствора уравнением Нернста. В потенциометрических измерениях широко применяются ионоселективные электроды, чувствительные преимущественно к какому-то одному иону в растворе: стеклянный электрод для измерения рН и электроды для селективного определения ионов натрия, аммония, фтора, кальция, магния и др. В поверхностный слой ионоселективного электрода могут быть включены ферменты, и в результате получается система, чувствительная к соответствующему субстрату. Отметим, что потенциал ионоселективного электрода определяется не переносом электронов, как в случае веществ с электронной проводимостью, а в основном переносом или обменом ионов. Однако уравнение Нернста, связывающее электродный потенциал с логарифмом концентрации (или активности) вещества в растворе, применимо и к такому электроду. При потенциометрическом титровании реагент добавляют в анализируемый раствор порциями и следят за изменением потенциала. S-образные кривые, характерные для такого типа титрования, позволяют определить точку эквивалентности и найти такие термодинамические параметры, как константа равновесия и стандартный потенциал.
1.4 Амперометрия
Метод основан на измерении предельного
диффузионного тока, проходящего через раствор при фиксированном напряжении
между индикаторным электродом и электродом сравнения. При амперометрическом
титровании точку эквивалентности определяют по излому кривой ток – объем
добавляемого рабочего раствора. Хроноамперометрические методы основаны на
измерении зависимости тока от времени и применяются в основном для определения
коэффициентов диффузии и констант скорости. По принципу амперометрии (как и
вольтамперометрии) работают миниатюрные электрохимические ячейки, служащие
датчиками на выходе колонок жидкостных хроматографов. Гальваностатические
методы аналогичны амперометрическим, но в них измеряется потенциал при
прохождении через ячейку тока определенной величины. Так, в хронопотенциометрии
контролируется изменение потенциала во времени. Эти методы применяются главным
образом для изучения кинетики электродных реакций.
1.5 Кулонометрия.
В кулонометрии при
контролируемом потенциале проводят полный электролиз раствора, интенсивно
перемешивая его в электролизере с относительно большим рабочим электродом
(донная ртуть или платиновая сетка). Полное количество электричества (Q, Кл), необходимое для электролиза, связано
с количеством образующего вещества (А, г) законом Фарадея:
где M – мол. масса (г/моль), F число Фарадея. Кулонометрическое титрование заключается в том, что при постоянном токе электролитически генерируют реактив, вступающий во взаимодействие с определяемым веществом. Ход титрования контролируют потенциометрически или амперометрически. Кулонометрические методы удобны тем, что являются по своей природе абсолютными (т.е. позволяют рассчитать количество определяемого вещества, не прибегая к калибровочным кривым) и нечувствительны к изменению условий электролиза и параметров электролизера (площади поверхности электрода или интенсивности перемешивания). При кулоногравиметрии количество вещества, подвергшегося электролизу, определяют взвешиванием электрода до и после электролиза.
Существуют и другие электроаналитические методы. В переменно-токовой полярографии на линейно меняющийся потенциал налагают синусоидальное напряжение малой амплитуды в широкой области частот и определяют либо амплитуду и фазовый сдвиг результирующего переменного тока, либо импеданс. Из этих данных получают информацию о природе веществ в растворе и о механизме и кинетике электродных реакций. В тонкослойных методах используются электрохимические ячейки со слоем электролита толщиной 10–100 мкм. В таких ячейках электролиз идет быстрее, чем в обычных электролизерах. Для изучения электродных процессов применяют спектрохимические методы со спектрофотометрической регистрацией. Для анализа веществ, образующихся на поверхности электрода, измеряют поглощение ими света в видимой, УФ- и ИК-областях. За изменением свойств поверхности электрода и среды следят с помощью методов электроотражения и эллипсометрии, которые основаны на измерении отражения излучения от поверхности электрода. К ним относятся методы зеркального отражения и комбинационного рассеяния света (рамановская спектроскопия), спектроскопия второй гармоники (фурье-спектроскопия).
1.6 Другие электрохимические явления и методы
При относительном движении электролита и заряженных частиц или поверхностей возникают электрокинетические эффекты. Важным примером такого рода является электрофорез, при котором происходит разделение заряженных частиц (например, молекул белка или коллоидных частиц), движущихся в электрическом поле. Электрофоретические методы широко используют для разделения белков или дезоксирибонуклеиновых кислот (ДНК) в геле. Электрические явления играют большую роль в функционировании живых организмов: они отвечают за генерацию и распространение нервных импульсов, возникновение трансмембранных потенциалов и т.д. Различные электрохимические методы применяются для изучения биологических систем и их компонентов. Представляет интерес и изучение действия света на электрохимические процессы. Так, предметом фотоэлектрохимических исследований являются генерация электрической энергии и инициация химических реакций под действием света, что весьма существенно для повышения эффективности преобразования солнечной энергии в электрическую. Здесь обычно используются полупроводниковые электроды из диоксида титана, сульфида кадмия, арсенида галлия и кремния. Еще одно интересное явление – электрохемилюминесценция, т.е. генерация света в электрохимической ячейке. Оно наблюдается, когда на электродах образуются высокоэнергетические продукты. Часто процесс проводят в циклическом режиме, чтобы получить как окисленную, так и восстановленную формы данного соединения. Взаимодействие их между собой приводит к образованию возбужденных молекул, которые переходят в основное состояние с испусканием света.
1.7 Прикладная электрохимия
Электрохимия имеет много практических применений. При помощи первичных гальванических элементов (элементов одноразового действия), соединенных в батареи, преобразуют химическую энергию в электрическую. Вторичные источники тока – аккумуляторы – запасают электрическую энергию. Топливные элементы – первичные источники тока, которые генерируют электричество благодаря непрерывной подаче реагирующих веществ (например, водорода и кислорода). Эти принципы лежат в основе портативных источников тока и аккумуляторов, применяющихся на космических станциях, в электромобилях и электронных приборах.
На электрохимическом синтезе основано крупнотоннажное производство многих веществ. При электролизе рассола в хлорщелочном процессе образуются хлор и щелочь, которые затем применяются для получения органических соединений и полимеров, а также в целлюлозно-бумажной промышленности. Продуктами электролиза являются такие соединения, как хлорат натрия, персульфат, перманганат натрия; электроэкстракцией получают важные в промышленном отношении металлы: алюминий, магний, литий, натрий и титан. В качестве электролитов лучше использовать расплавы солей, поскольку в этом случае, в отличие от водных растворов, восстановление металлов не осложняется выделением водорода. Электролизом в расплаве соли получают фтор. Электрохимические процессы служат основной для синтеза некоторых органических соединений; например, гидродимеризацией акрилонитрила получают адипонитрил (полупродукт в синтезе найлона).
Широко практикуется нанесение на различные предметы гальванических покрытий из серебра, золота, хрома, латуни, бронзы и других металлов и сплавов с целью защиты изделий из стали от коррозии, в декоративных целях, для изготовления электрических разъемов и печатных плат в электронной промышленности. Электрохимические методы используются для высокоточной размерной обработки заготовок из металлов и сплавов, особенно таких, которые не удается обрабатывать обычными механическими способами, а также для изготовления деталей сложного профиля. При анодировании поверхности таких металлов, как алюминий и титан, образуются защитные оксидные пленки. Такие пленки создают на поверхности заготовок из алюминия, тантала и ниобия при изготовлении электролитических конденсаторов, а иногда в декоративных целях.
Кроме того, на электрохимических методах часто базируются исследования коррозионных процессов и подбор материалов, замедляющих эти процессы. Коррозию металлических конструкций можно предотвратить с помощью катодной защиты, для чего внешний источник подсоединяют к защищаемой конструкции и аноду и поддерживают такой потенциал конструкции, при котором ее окисление исключается. Исследуются возможности практического применения других электрохимических процессов. Так, для очистки воды можно использовать электролиз. Весьма перспективное направление – преобразование солнечной энергии с помощью фотохимических методов. Разрабатываются электрохимические мониторы, принцип действия которых основан на электрохемилюминесценции.
Электрохимические методы анализа (электроанализ), в основе которых лежат электрохимические процессы, занимают достойное место среди методов контроля состояния окружающей среды, так как способны обеспечить определение огромного числа как неорганических, так и органических экологически опасных веществ. Для них характерны высокая чувствительность и селективность, быстрота отклика на изменение состава анализируемого объекта, легкость автоматизации и возможность дистанционного управления. И наконец, они не требуют дорогостоящего аналитического оборудования и могут применяться в лабораторных, производственных и полевых условиях. Непосредственное отношение к рассматриваемой проблеме имеют три электроаналитических метода: вольтамперометрия, кулонометрия и потенциометрия.
Краткая историческая справка . Начало развития электроанализа связывают с возникновением классического электрогравиметрического метода (около 1864 года, У. Гиббс). Открытие М. Фарадеем в 1834 году законов электролиза легло в основу метода кулонометрии, однако применение этого метода началось с 30-х годов ХХ века. Настоящий перелом в развитии электроанализа произошел после открытия в 1922 году Я. Гейровским метода полярографии. Полярографию можно определить как электролиз с капающим ртутным электродом. Этот метод остается одним из основных методов аналитической химии. В конце 50-х - начале 60-х годов проблема охраны окружающей среды стимулировала бурное развитие аналитической химии, и в частности электроаналитической химии, включая полярографию. В результате были разработаны усовершенствованные полярографические методы: переменнотоковая (г. Баркер, Б. Брейер) и импульсная полярография (г. Баркср, А. Гарднср), которые значительно превосходили по своим характеристикам классический вариант полярографии, предложенный Я. Гейровским. При использовании твердых электродов из различных материалов вместо ртутных (используемых в полярографии) соотвстствуюшие методы стали называться вольтамперометрическими. В конце 50-х годов работы В. Кемули и 3. Кублика положили начало методу инверсионной вольтамперометрии. Наряду с методами кулонометрии и вольтамперометрии развиваются методы, основанные на измерении электродных потенциалов и электродвижущих сил гальванических элементов, - методы потенциометрии и ионометрии (см. ).
Вольтамперометрия
. Это группа методов, основанных на
изучении зависимости силы тока в электролитической ячейке от величины
потенциала, приложенного к погруженному в анализируемый раствор индикаторному
микроэлектроду. Эти методы основаны на принципах электролиза; присутствующие в
растворе определяемые вещества окисляются или восстанавливаются на индикаторном
электроде. В ячейку помещают помимо индикаторного еще электрод сравнения со
значительно большей поверхностью, чтобы при прохождении тока его потенциал
практически не менялся. В качестве индикаторных микроэлектродов наиболее часто
используют стационарные и вращающиеся электроды из платины или графита, а также
ртутный капающий электрод, представляющий собой длинный узкий капилляр, на
конце которого периодически образуются и отрываются небольшие ртутные капли
диаметром 1-2 мм (рис. 1). Качественный и количественный составы раствора могут
быть установлены из вольтамперограмм.
Рис. 4. Электрохимическая ячейка с
капающим ртутным электродом: 1 - анализируемый раствор, 2 - ртутный капающий
электрод, 3 - резервуар с ртутью, 4 - электрод сравнения
Вольтамперометрические методы, особенно такие чувствительные варианты, как дифференциальная импульсная полярография и инверсионная вольтамперометрия, постоянно используются во всех областях химического анализа и наиболее полезны при решении проблем охраны окружающей среды. Эти методы применимы для определения и органических и неорганических веществ, например для определения большинства химических элементов. С помощью метода инверсионной вольтамперометрии чаще всего решают проблему определения следов тяжелых металлов в водах и биологических материалах. Так, например, вольтамперометрические методики одновременного определения Си, Cd и РЬ, а также Zn и РЬ или ТI в питьевой воде включены в стандарт ФРГ. Важным достоинством вольтамперометрии является возможность идентифицировать формы нахождения ионов металлов в водах. Это позволяет оценивать качество воды, так как разные химические формы существования металлов обладают разной степенью токсичности. Из органических веществ можно определять соединения, обладающие группами, способными к восстановлению (альдегиды, кетоны, нитро -, нитрозосоединения, ненасыщенные соединения, галогенсодержащие соединения, азосоединения) или окислению (ароматические углеводороды, амины, фенолы, алифатические кислоты, спирты, серусодержашие соединения). Возможности определения органических вешеств методом инверсионной вольтамперометрии существенно расширяются при использовании химически модифицированных электродов. Модификацией поверхности электрода полимерными и неорганическими пленками, включаюшими реагенты со специфическими функциональными группами, в том числе и биомолекулы, можно создать для определяемого компонента такие условия, когда аналитический сигнал будет практически специфичным. Использование модифицированных электродов обеспечивает избирательное определение соединений с близкими окислительно-восстановительными свойствами (например, пестицидов и их метаболитов) или электрохимически неактивных на обычных электродах. Вольтамперометрию применяют для анализа растворов, но она может быть использована и для анализа газов. Сконструировано множество простых вольтамперометрических анализаторов для работы в полевых условиях.
Кулонометрия . Метод анализа, основанный на измерении количества электричества (Q), прошедшего через электролизер при электрохимическом окислении или восстановлении вещества на рабочем электроде. Согласно закону Фарадея, масса электрохимически превращенного вещества (Р) связана с Q соотношением:
P
=
QM
/
Fn
,
где М - молекулярная или атомная масса вещества, п - число электронов, вовлеченных в электрохимическое превращение одной молекулы (атома) вещества, р - постоянная Фарадея.
Различают прямую кулонометрию и кулонометрическос титрование. В первом случае определяют электрохимически активное вещество, которое осаждают (или переводят в новую степень окисления) на электроде при заданном потенциале электролиза, при этом затраченное количество электричества пропорционально количеству прореагировавшего вещества. Во втором случае в анализируемый раствор вводят электрохимически активный вспомогательный реагент, из которого электролитически генерируют титрант (кулонометрический титрант), и он количественно химически взаимодействует с определяемым веществом. Содержание определяемого компонента оценивают по количеству электричества, прошедшего через раствор при генерировании титранта вплоть до момента завершения химической реакции, который устанавливают, например, с помощью цветных индикаторов. Важно, чтобы при проведении кулонометрического анализа в исследуемом растворе отсутствовали посторонние вещества, способные вступать в электрохимические или химические реакции в тех же условиях, то есть не протекали побочные электрохимические и химические процессы.
Кулонометрию используют для определения как следовых (на уровне 109-10 R моль/л), так и весьма больших количеств веществ с высокой точностью. Кулонометрически можно определять многие неорганические (практически все металлы, в том числе тяжелые, галогены, S, NО з, N0 2) и органические вещества (ароматические амины, нитро- и нитрозосоединения, фенолы, азокрасители). Автоматические кулонометрические анализаторы для определения очень низких содержаний (до 104 %) газообразных загрязнений (S02" Оз, H 2 S, NO, N0 2) в атмосфере успешно зарекомендовали себя в полевых условиях.
Потенциометрия. Метод анализа, основанный на зависимости paвновесного электродного потенциала Е от активности а компонентов электрохимической реакции: аА + ЬВ + пе = тМ + рР.
При потенциометрических измерениях составляют гальванический элемент из индикаторного электрода, потенциал которого зависит от активности одного из компонентов раствора, и электрода сравнения и измеряют электродвижущую силу этого элемента.
Различают прямую потенциометрию и потенциометрическое титрование. Прямая потенциометрия применяется для непосредственного определения активности ионов по значению потенциала (Е) соответствующего индикаторного электрода. В методе потенциометрического титрования регистрируют изменение Е в ходе реакции определяемого компонента с подходящим титрантом.
При решении задач охраны окружающей среды наиболее важен метод прямой потенциометрии с использованием мембранных ионоселективных электродов (ИСЭ) - ионометрия. В отличие от многих других методов анализа, позволяющих оценить лишь общую концентрацию веществ, ионометрия позволяет оценить активность свободных ионов и поэтому играет большую роль в изучении распределения ионов между их различными химическими формами. Для контроля объектов окружающей среды особенно важны методы автоматизированного мониторинга, и использование ИСЭ очень удобно для этой цели.
Одним из основных показателей при характеристике состояния окружающей среды является значение рН среды, определение которого обычно проводят с помощью стеклянных электродов. Стеклянные электроды, покрытые полупроницаемой мембраной с пленкой соответствующего электролита, используют в анализе вод и атмосферы для контроля загрязнений (NН з, SO 2 NO, NO 2 , СO 2 , H 2 S). ИСЭ применяют обычно при контроле содержания анионов, для которых методов определения традиционно значительно меньше, чем для катионов. К настоящему времени разработаны и повсеместно применяются ИСЭ для определения F, СI , Вг, I , С1O 4 , CN , S 2 , NO] и NO 2 , позволяющие определять перечисленные ионы в интервале концентраций от 10 -6 до 10 -1 моль/л.
Одной из важных областей применения ионометрии являются гидрохимические исследования и определение концентрации анионов и катионов в разных типах вод (поверхностных, морских, дождевых). Другая область применения ИСЭ - анализ пищевых продуктов. Примером может служить определение NO – 3 и NO 2 - в овощах, мясных и молочных продуктах, продуктах детского питания. Создан миниатюрный ИСЭ в форме иглы для определения NO - 3 непосредственно в мякоти плодов и овощей.
Широко используется ионометрия и для определения различных биологически активных соединений и лекарственных препаратов. В настоящее время уже можно говорить, что существуют носители, селективные практически к любому типу органических соединений, а это означает что возможно создание неограниченного числа соответствующих ИСЭ. Перспективным направлением является использование ферментных электродов, в мембрану которых включены иммобилизованные ферменты. Эти электроды обладают высокой специфичностью, свойственной ферментативным реакциям. С их помощью, например, удастся определять ингибирующие холинэстеразу, инсектициды (фосфорорганические соединения, карбаматы) при концентрациях -1 нг/мл. Будущее метода связано с созданием компактных специфичных сенсоров, представляющих собой современные электронные устройства в cочетании с ионоселективными мембранами, которые позволят обходиться без разделения компонентов проб и заметно ускорят проведение анализов в полевых условиях.
Анализ сточных вод
Электроаналитические методы, которые обычно применяют в анализе воды для определения неорганических компонентов, часто уступают по чувствительности методам газовой и жидкостной хроматографии, атомно-адсорбционной спектрометрии. Однако здесь используется более дешевая аппаратура, иногда даже в полевых условиях. Основными электроаналитическими методами, применяемыми в анализе воды, являются вольтамперометрия, потенциометрия и кондуктометрия. Наиболее эффективными вольтамперометрическими методами являются дифференциальная импульсная полярография (ДИП) и инверсионный электрохимический анализ (ИЭА). Сочетание этих двух методов позволяет проводить определение с очень высокой чувствительностью - приблизительно 10 -9 моль/л, аппаратурное оформление при этом несложно, что дает возможность делать анализы в полевых условиях. На принципе использования метода ИЭА или сочетания ИЭА с ДИП работают полностью автоматизированные станции мониторинга. Методы ДИП и ИЭА в прямом варианте, а также в сочетании друг с другом используют для анализа загрязненности воды ионами тяжелых металлов, различными органическими веществами. При этом часто способы пробоподготовки являются гораздо более простыми, чем в спектрометрии или газовой хроматографии. Преимуществом метода ИЭА является (в отличие от других методов, например, атомно-адсорбционной спектрометрии) также способность “отличать” свободные ионы от их связанных химических форм, что важно и для оценки физико-химических свойств анализируемых веществ, и с точки зрения биологического контроля (например, при оценке токсичности вод). Время проведения анализа иногда сокращается до нескольких секунд за счет повышения скорости развертки поляризующего напряжения.
Потенциометрия с применением различных ионоселективных электродов используется в анализе воды для определения большого числа неорганических катионов и анионов. Концентрации, которые удается определить таким способом, 10 0 -10 -7 моль/л. Контроль с помощью ионоселективных электродов отличается простотой, экспрессностью и возможностью проведения непрерывных измерений. В настоящее время созданы ионоселективные электроды, чувствительные к некоторым органическим веществам (например, алкалоидам), поверхностно-активным веществами и моющим веществам (детергентам). В анализе воды используются компактные анализаторы типа зондов с применением современных ионоселективных электродов. При этом в ручке зонда смонтирована схема, обрабатывающая отклик, и дисплей.
Кондуктометрия
используется в работе анализаторов
детергентов в сточных водах, при определении концентраций синтетических
удобрений в оросительных системах, при оценке качества питьевой воды. В
дополнение к прямой кондуктометрии для определения некоторых видов
загрязнителей могут быть использованы косвенные методы, в которых определяемые
вещества взаимодействуют перед измерением со специально подобранными реагентами
и регистрируемое изменение электропроводности вызывается только присутствием
соответствующих продуктов реакции. Кроме классических вариантов кондуктометрии
применяют и ее высокочастотный вариант (осциллометрию), в котором индикаторная
электродная система реализуется в кондуктометрических анализаторах непрерывного
действия.
Глава
3. Приборы на основе электрохимических методов анализа
Вольтамперометрический метод анализа сегодня считается одним из наиболее перспективных среди электрохимических методов, благодаря его широким возможностям и хорошим эксплутационным характеристикам.
Современная инверсионная вольтамперометрия, заменившая классическую полярографию, - высокочувствительный и экспрессный метод определения широкого круга неорганических и органических веществ, обладающих окислительно-восстановительными свойствами.
Это один из наиболее универсальных методов определения следовых количеств веществ, который с успехом применяется для анализа природных гео- и биологических, а также медицинских, фармацевтических и иных объектов.
Вольтамперометрические анализаторы делают возможным одновременное определение нескольких компонентов (до 4 - 5) в одной пробе с довольно высокой чувствительностью 10 -8 - 10 -2 М (а инверсионная вольтамперометрия - до 10-10 - 10 -9 М).
Наиболее перспективной в аналитической химии сегодня считается адсорбционная инверсионная вольтамперометрия, основанная на предварительном адсорбционном концентрировании определяемого элемента на поверхности электрода и последующей регистрации вольтамперограммы полученного продукта. Таким образом можно концентрировать многие органические вещества, а также ионы металлов в виде комплексов с органическими лигандами (особенно азот - и серусодержащими). При времени последовательного накопления 60 с и использовании дифференциального импульсного режима регистрации вольтамперограммы удается достичь пределов обнаружения на уровне 10 -10 - 10 -11 моль/л (10 -8 - 10 -9 г/л или 0,01 - 0,001 мкг/дм 3).
Вольтамперометрический комплекс анализа металлов «ИВА - 400МК» (НПКФ »Аквилон», Москва) предназначен для анализа 30 элементов (Cu, Zn, Pb, Cd, As, Co, Ni, Cr, и др. металлы), чувствительность 0,1 - 10 -3 мкг/дм 3 .
Вольтамперометрический анализатор с УФ-облучением проб - ТА-1М (Томск) , который, помимо ионов металлов, позволяет определять целый ряд органических соединений. Для прибора характерны следующие особенности:
· одновременный анализ в трех электрохимических ячейках,
· малая навеска пробы (0,1 - 1,0 г),
· низкая стоимость пробоподготовки и анализа.
В Санкт – Перебурге НФТ «Вольта» выпускает вольтамперометрический комплекс «АВС-1» с вращающимся дисковым стеклоуглеродным электродом, который позволяет проводить анализ токсичных элементов в водах, пищевых продуктах и различных материалах. Предел обнаружения без концентрирования пробы составляет: 0,1 мг/л для Pb, 0,5 мг/л для Cd, 1,0 мкг/л для Cu. Объем пробы - 20 мл, время получения вольтамперной кривой не более 3 мин.
«АЖЭ - 12» (Владикавказ ) предназначен для экспресс-анализа ионного состава сточных и оборотных вод. В анализаторе используется традиционный ртутный электрод. Контролируемые компоненты - Cu, Zn, Pb, Cd, In, Bi, Tl, Sb, As, Co, Ni, Cr, CN - , Cl - , S 2- . Анализатор позволяет проводить измерения без пробоподготовки.
«Экотест-ВА» («Эконикс», Москва ) - портативный вольтамперометрический анализатор. Выполнен на современной микропроцессорной элементной базе и оснащен целым комплексом электродов - графитовым, стеклоуглеродным, микроэлектродами из благородных металлов и ртутным капающим электродом.
Приборы этой серии предназначены для определения металлов Cu, Zn, Pb, Cd, As, Bi, Mn, Co, Ni, Cr, а также ацетальдегида, фурфурола, капролактама и др. веществ в пробах питьевой, природной, сточной воды, почве, а после соответствующей пробоподготовки - в пищевых продуктах и кормах.
Возможности многих аналитических методов анализа вод могут значительно расшириться при применении в процессе пробоподготовки проточно-инжекционных концентрирующих приставок, работающих в автоматическом режиме - например, типа БПИ-М и БПИ-Н.
БПИ-М - предназначен для автоматизированной пробоподготовки, в его состав входят микроколонки с высокоэффективными сорбентами. Производительность блока - 30-60 анализов в день при полной автоматизации процесса. Применение блока позволяет повысить чувствительность в 20 раз за минуту концентрирования. Блок наиболее хорошо работает в сочетании с атомно-абсорбционным детектированием, а также с рентгено-флуоресцентным, атомно-абсорбционным и электрохимическими методами.
БПИ-Н - предназначен для концентрирования ионов металлов на избирательных сорбентах одновременно в четырех микроколонках с ДЭТАТА - сорбентом или на 4 тонкослойных сорбционных ДЭТАТА - фильтрах. Возможно его использование с рентгено-флуоресцентным, атомно-абсорбционным, атомно-эмиссионным, электрохимическим методами.
Анализаторы на основе вольтамперометрии
Приборы на принципе инверсной вольтамперометрии пользуются в последнее время особым спросом. В них селективность и высокая чувствительность сочетаются с простотой анализа.
В отношении определения элементного состава (например, по тяжелым металлам) эти приборы успешно конкурируют с атомно-абсорбционными спектрофотометрами, так как не уступают им по чувствительности, но значительно более компактны и дешевы (примерно в 5 - 10 раз). Они не требуют дополнительных расходных материалов, а также дают возможность одновременного экспрессного определения нескольких элементов.
Полярограф АВС - 1.1 (НТФ «Вольта» Спб).
Пределы обнаружения металлов без концентрирования пробы составляют (мг/л): Cd, Pb, Bi - 0,0001, Hg - 0,00015, Cu - 0,0005, Zn, Ni - 0,01. Стоимость 1700$.
Анализаторы на кондуктометрическом принципе предназначены для количественного определения суммарного содержания солей в воде. «ЭКА-2М» (Санкт-Петербург) измеряет солесодержание в широком интервале значений от 0,05 до 1000 мкСм/см (900$). «АНИОН», «МАРК», КСЛ (от 330 до 900 $), ХПК - анализаторы (750 $).
Газоанализаторы вредных веществ
Автоматический газоанализатор представляет собой прибор, в котором отбор проб воздуха, определение количества контролируемого компонента, выдача и запись результатов анализа проводится автоматически по заданной программе без участия оператора. Для контроля воздушной среды используют газоанализаторы, работа которых основана на различных принципах.
Термокондуктометрические газоанализаторы.
Принцип работы основан на зависимости теплопроводности газовой смеси от ее состава. Чувствительным элементом анализаторов этого типа являются тонкие платиновые нити. В зависимости от состава газа меняется температура чувствительного элемента, возникает ток, сила которого пропорциональна концентрации контролируемого компонента.
Кулонометрические газоанализаторы .
Принцип работы основан на измерении предельного электрического тока, возникающего при электролизе раствора, который содержит определяемое вещество, являющееся электрохимическим деполяризатором. Анализируемая смесь, содержащая, например, диоксид серы, подается в электрохимическую ячейку. Он реагирует с иодом до образования сероводорода, который затем электороокисляется на измерительном электроде. Электрический ток является мерой концентрации определяемого компонента.
ГЛАВА 4.
ОБЗОР
WEB
–САЙТОВ ФИРМ–ПРОДАВЦОВ ХИМИКО –
АНАЛИТИЧЕСКОГО ОБОРУДОВАНИЯ
"AGILENT.RU"
Современное тестовое, измерительное и мониторинговое оборудование для разработки, изготовления и внедрения новых электронных приборов и технологий...
http://www.agilent.ru
"АКАДЕМЛАЙН", ЗАО, Москва
Поставляет широкую номенклатуру измерительного химико-аналитического оборудования...
http://www.academline.com/
"АКТАКОМ"
Зарегистрированная торговая марка АКТАКОМ объединяет в себе широкий спектр контрольно-измерительной аппаратуры мирового класса. Все лучшее от зарубежных и отечественных производителей...
http://www.aktakom.ru
"АНАЛИТПРИБОР"
Предлагает газоанализаторы
http://www.analytpribor.ru
"ВАТСОН", АО, Мытищи Московской обл.
Приборы и средства измерений;
http://www.watson.ru/
"ДИПОЛЬ", НПФ, Санкт-Петербург
http://www.dipaul.ru/
"ЕвроЛаб СПб", ООО, Санкт-Петербург
Приборы спектрального анализа, хроматографы.
http://www.eurolab.ru
"IZME.RU"
http://www.izme.ru/
"ИНСОВТ", ЗАО
Разработка и производство газоанализаторов
http://www.insovt.ru
"Институт информационных технологий", Минск, Беларусь
Специализируется на разработке и производстве измерительных приборов для волоконной оптики...
"КИПАРИС", ООО, Санкт-Петербург
http://www.kiparis.spb.ru/
"КОНТИНЕНТ", Гомель
http://www.continent.h1.ru
"Контрольно-измерительные приборы и оборудование", Волгоград
http://www.oscilloscop.ru
"Контур", ИТЦ, ООО, Новосибирск
http://www.kip.ru/
"КрайСибСтрой", ООО, Красноярск
http://www.kipkr.ru/
"Крисмас+", ЗАО, Санкт-Петербург
http://www.christmas-plus.ru
"КУРС", ООО, Санкт-Петербург
http://www.kypc.spb.ru
"ЛЮМЭКС", Санкт-Петербург
http://www.lumex.ru/
"МЕТТЕК"
http://www.mettek.ru
"МЕТТЛЕР ТОЛЕДО"
http://www.mt.com
"МОНИТОРИНГ", НТЦ, Санкт-Петербург
http://www.monitoring.vniim.ru
"Научные приборы", ОАО, Санкт-Петербург
http://www.sinstr.ru
"НеваЛаб", ЗАО, Санкт-Петербург
http://www.nevalab.ru
"ОВЕН", ПО, Москва
http://www.owen.ru/
"ОКТАВА+", Москва
http://www.octava.ru/
"ОПТЭК", ЗАО, Санкт-Петербург
Разрабатывает и производит газоанализаторы и аналитические системы различного назначения для использования в экологии, промышленности и научных исследованиях...
http://www.optec.ru
"ПОЛИТЕХФОРМ", Москва
http://www.ptfm.ru
"Практик-НЦ", ОАО, Москва, Зеленоград
http://www.pnc.ru/
"ПРИБОРЫ И АНАЛИТИЧЕСКАЯ ТЕХНИКА"
Приборы для химического анализа.
http://www.zhdanov.ru/
"Сартогосм", ЗАО, Санкт-Петербург
http://www.sartogosm.ru
"Специал", ЗАО, Москва
http://www.special.ru
"ТКА"
http://www.tka.spb.ru/
"ТСТ", ЗАО, Санкт-Петербург
http://www.tst-spb.ru
"ЭКОПРИБОР", НПО, Москва
Предлагает газоанализаторы и газоаналитические системы...
http://ecopribor.ru
"ЭКОТЕХ", МСП, Украина
http://ecotech.dn.ua
"ЭКОТЕХИНВЕСТ", НПФ, Москва
http://ecotechinvest.webzone.ru
"Эксис", ЗАО, Москва, Зеленоград
http://www.eksis.ru/
"ЭЛИКС"
http://www.eliks.ru/
"ЭМИ", ООО, Санкт-Петербург
Производство оптических газоанализаторов, анализаторов нефтепродуктов.
http://www.igm.spb.ru
"ЭНЕРГОТЕСТ", ЗАО, Москва
http://www.energotest.ru, http://www.eneffect.ru
ХИММЕД
Аналитические приборы и хроматография
е -mail: [email protected]
ЛИТЕРАТУРА
1. Гейровский Я., Кута Я., Основы полярографии, пер. с чеш., М., 1965;
2. Га л юс 3., Теоретические основы электрохимического анализа, пер. с польск., М., 1974;
3. Каплан Б. Я., Импульсная полярография, М., 1978;
4. Брайнина X. 3., Нейман Е. Я., Твердофазные реакции в электроаналитической химии, М., 1982;
5. Каплан Б. Я., Пац Р. Г., Салихджанова Р. М.-Ф., Вольтамперометрия переменного тока, М., 1985.
6. Плэмбек Дж. Электрохимические методы анализа. / Пер. с англ. М.: Мир, 1985. 496 с.
7. Краткая химическая энциклопедия. М.: Советская энциклопедия, 1964. Том 1. А–Е. 758 c.
8. Классификация и номенклатура электрохимических методов // Журн. аналит. химии. 1978. Т. 33, вып. 8. С. 1647–1665.
9. Recommended Terms, Symbols and Definitions for Electroanalytical Chemistry // Pure & Appl. Chem. 1979. Vol. 51. P. 1159–1174.
10. Об использовании понятия «химический эквивалент» и связанных с ним величин: Журн. аналит. химии. 1989. Т. 44, вып. 4. С. 762–764; Журн. аналит. химии. 1982. Т. 37, вып. 5. С. 946; Журн. аналит. химии. 1982. Т. 37, вып. 5. С. 947.
11. Нейман Е.Я. Терминология современной аналитической химии и ее формирование // Журн. аналит. химии. 1991. Т. 46, вып. 2. С. 393–405.
12. Представление результатов химического анализа (Рекомендации IUPAC 1994 г.) // Журн. аналит. химии. 1998. Т. 53. № 9. С. 999–1008.
13. Compendium of Analytical Nomenclature (Definitive Rules 1997). 3rd ed., IUPAC, Blackwell Science, 1998. 8.1–8.51 (Electrochemical Analysis).
Введение
Применение электрохимических методов в количественном анализе базируется на использовании зависимостей величин измеряемых параметров электрохимических процессов (разности электрических потенциалов, тока, количества электричества) от содержания определяемого вещества в анализируемом растворе, участвующего в данном электрохимическом процессе. Электрохимические процессы - процессы, которые сопровождаются одновременным протеканием химических реакций и изменением электрических свойств системы, которую в подобных случаях можно назвать электрохимической системой. В аналитической практике электрохимическая система обычно содержит электрохимическую ячейку, включающую сосуд с электропроводящим анализируемым раствором, в который погружены электроды.
Классификация электрохимических методов анализа
Электрохимические методы анализа классифицируют по-разному. . Классификация, основанная на учете природы источника электрической энергии в системе. Различают две группы методов. -Методы без наложения внешнего (постороннего) потенциала. Источником электрической энергии служит сама электрохимическая система, представляющая собой гальванический элемент (гальваническую цепь). К таким методам относятся потенциометрические методы; электродвижущая сила (ЭДС) и электродные потенциалы в такой системе зависят от содержания определяемого вещества в растворе. - Методы с наложением внешнего (постороннего) потенциала. К таким методам относятся:
о кондуктометрический анализ - основан на измерении электрической проводимости растворов как функции их концентрации;
о вольтамперометрический анализ - основан на измерении тока как функции приложенной известной разности потенциалов и концентрации раствора;
о кулонометрический анализ - основан на измерении количества электричества, прошедшего через раствор, как функции его концентрации;
о электрогравиметрический анализ - основан на измерении массы продукта электрохимической реакции.
Классификация по способу применения электрохимических методов. Различают прямые и косвенные методы.
- Прямые методы. Измеряют электрохимический параметр как известную функцию концентрации раствора и по показанию соответствующего измерительного прибора находят содержание определяемого вещества в растворе.
- Косвенные методы. Методы титрования, в которых окончание титрования фиксируют на основании измерения электрических параметров системы.
В соответствии с данной классификацией различают, например, прямую кондуктометрию и кондуктометрическое титрование, прямую потенциометрию и потенциометрическое титрование и т.д.
В данном пособии приведены лабораторные работы только по следующим электрохимическим методам:
Прямой потенциометрией;
Потенциометрическому титрованию;
Кулонометрическому титрованию.
Все эти методы - фармакопейные и применяются для контроля качества лекарственных средств.
Общая характеристика потенциометрического анализа
Принцип метода
Потенциометрический анализ (потенциометрия) основан на измерении ЭДС и электродных потенциалов как функции концентрации анализируемого раствора.
Если в электрохимической системе - в гальваническом элементе - на электродах протекает реакция:
с переносом n электронов, то уравнение Нернста для ЭДС E этой реакции имеет вид:
где- стандартная ЭДС реакции (разность стандартных электродных потенциалов); R - универсальная газовая постоянная; T - абсолютная температура, при которой протекает реакция; F - число Фарадея;-
активности реагентов - участников реакции.
Уравнение (1) справедливо для ЭДС обратимо работающего гальванического элемента.
Для комнатной температуры уравнение (1) можно представить в форме:
 (2)
(2)
В условиях, когда активность реагентов приблизительно равна их концентрации, уравнение (1) переходит в уравнение (3):
 (3)
(3)
где - концентрации реагентов.
Для комнатной температуры это уравнение можно представить в виде:
 (4)
(4)
При потенциометрических измерениях в электрохимической ячейке используют два электрода:
. индикаторный электрод, потенциал которого зависит от концентрации определяемого (потенциалопределяющего) вещества в анализируемом растворе;
. электрод сравнения, потенциал которого в условиях проведения анализа остается постоянным.
Именно поэтому величину ЭДС, определяемую уравнениями (14), можно рассчитать как разность реальных потенциалов этих двух электродов.
В потенциометрии используют электроды следующих типов: электроды первого, второго рода, окислительно-восстановительные, мембранные.
Электроды первого рода. Это электроды, обратимые по катиону, общему с материалом электрода. Различают три разновидности электродов первого рода:
а) Металл M, погруженный в раствор соли того же металла. На поверхности таких электродов протекает обратимая реакция:

Реальный потенциал такого электрода первого рода зависит от активности катионов металла и описывается уравнениями (5-8). В общем случае для любой температуры:
катионов металла и описывается уравнениями (5-8). В общем случае для любой температуры:
 (5)
(5)
Для комнатной температуры:
 (6)
(6)
При малых концентрациях  , когда активность
, когда активность  катионов
катионов
металла приблизительно равна их концентрации,
 (7)
(7)
Для комнатной температуры:
 (8)
(8)
б) Газовые электроды, например, водородный электрод, в том числе и стандартный водородный электрод. Потенциал обратимо работающего газового водородного электрода определяется активностью ионов водорода, т.е. величиной рН раствора, и при комнатной температуре равен:
поскольку для водородного электрода стандартный потенциал принимается равным нулю  , а в соответствии с электродной реакцией
, а в соответствии с электродной реакцией

число электронов, участвующих в этой реакции, равно единице: n = 1;
в) Амальгамные электроды, представляющие собой амальгаму металла, погруженную в раствор, содержащий катионы того же металла. Потен-
циал таких электродов первого рода зависит от активности ка-
ка-
тионов металла в растворе и активности a(M) металла в амальгаме:

Амальгамные электроды обладают высокой обратимостью. Электроды второго рода обратимы по аниону. Различают следующие виды электродов второго рода:
А. Металл, поверхность которого покрыта малорастворимой солью этого же металла, погруженный в раствор, содержащий анионы, входящие в состав этой малорастворимой соли.
Примером могут служить хлорсеребряный электрод  , или каломельный электрод
, или каломельный электрод  ,
,

Хлорсеребряный электрод состоит из серебряной проволоки, покрытой малорастворимой в воде солью , погруженной в водный раствор хлорида калия. На хлорсеребряном электроде протекает обратимая реакция:

Каломельный электрод состоит из металлической ртути, покрытой пастой малорастворимого хлорида ртути(I) - каломели, контак-
- каломели, контак-
тирующей с водным раствором хлорида калия. На каломельном электроде протекает обратимая реакция:

Реальный потенциал электродов второго рода зависит от активности анионов и для обратимо работающего электрода, на котором протекает реакция

описывается уравнениями Нернста (9-12).
В общем случае при любой приемлемой температуре T:
 . (9)
. (9)
Для комнатной температуры:
Для условий, в которых активность анионов приблизительно равна их концентрации :
:
 . (11)
. (11)
Для комнатной температуры:
 (12)
(12)
Например, реальные потенциалыисоответственно хлорсеребряного и каломельного электродов при комнатной температуре можно представить в виде:

В последнем случае в электродной реакции участвуют 2 электрона (n = 2) и образуются также 2 хлорид-иона, поэтому множитель при логарифме равен также 0,059.
Электроды второго рода рассмотренного вида обладают высокой обратимостью и стабильны в работе, поэтому их часто используют в качестве электродов сравнения, способных устойчиво поддерживать постоянное значение потенциала;
б) газовые электроды второго рода, например, хлорный электрод
, Газовые электроды второго рода в количественном потен-
Газовые электроды второго рода в количественном потен-
циометрическом анализе применяются редко.
Окислительно-восстановительные электроды. Состоят из инертного материала (платины, золота, вольфрама, титана, графита и др.), погруженного в раствор, содержащий окисленную Ox и восстановленную Red формы данного вещества. Существуют две разновидности окислительновосстановительных электродов:
1) электроды, потенциал которых не зависит от активности ионов водорода, например и т.д.;
2) электроды, потенциал которых зависит от активности ионов водорода, например, хингидронный электрод.
На окислительно-восстановительном электроде, потенциал которого не зависит от активности ионов водорода, протекает обратимая реакция:

Реальный потенциал такого окислительно-восстановительного электрода зависит от активности окисленной и восстановленной формы данного вещества и для обратимо работающего электрода описывается, в зависимости от условий (по аналогии с вышерассмотренными потенциалами), уравнениями Нернста (13-16):
(13) (14) (15)  (16)
(16)
где все обозначения - традиционные.
Если в электродной реакции участвуют ионы водорода, то их активность (концентрацию) учитывают в соответствующих уравнениях Нернста для каждого конкретного случая.
Мембранные, или ион-селективные, электроды - электроды, обратимые по тем или иным ионам (катионам или анионам), сорбируемым твердой или жидкой мембраной. Реальный потенциал таких электродов зависит от активности тех ионов в растворе, которые сорбируются мембраной.
Мембранные электроды с твердой мембраной содержат очень тонкую мембрану, по обе стороны которой находятся разные растворы, содержащие одни и те же определяемые ионы, но с неодинаковой концентрацией: раствор (стандартный) с точно известной концентрацией определяемых ионов и анализируемый раствор с неизвестной концентрацией определяемых ионов. Вследствие различной концентрации ионов в обоих растворах ионы на разных сторонах мембраны сорбируются в неодинаковых количествах, неодинаков и возникающий при сорбции ионов электрический заряд на разных сторонах мембраны. Как результат возникает мембранная разность потенциалов.
Определение ионов с применением мембранных ион-селективных электродов называют ионометрией.
Как уже говорилось выше, при потенциометрических измерениях электрохимическая ячейка включает два электрода - индикаторный
и электрод сравнения. Величина ЭДС, генерируемой в ячейке, равна разности потенциалов этих двух электродов. Поскольку потенциал электрода сравнения в условиях проведения потенциометрического определения остается постоянным, ЭДС зависит только от потенциала индикаторного электрода, т.е. от активности (концентрации) тех или иных ионов в растворе. На этом и основано потенциометрическое определение концентрации данного вещества в анализируемом растворе.
Для потенциометрического определения концентрации вещества в растворе применяют как прямую потенциометрию, так и потенциометрическое титрование, хотя второй способ используют намного чаще первого.
Прямая потенциометрия
Определение концентрации вещества в прямой потенциометрии. Проводят обычно методом градуировочного графика или методом добавок стандарта.
. Метод градуировочного графика. Готовят серию из 5-7 эталонных растворов с известным содержанием определяемого вещества. Концентрация определяемого вещества и ионная сила в эталонных растворах не должны сильно отличаться от концентрации и ионной силы анализируемого раствора: в этих условиях уменьшаются ошибки определения. Ионную силу всех растворов поддерживают постоянной введением индифферентного электролита. Эталонные растворы последовательно вносят в электрохимическую (потенциометрическую) ячейку. Обычно эта ячейка представляет собой стеклянный химический стакан, в который помещают индикаторный электрод и электрод сравнения.
Измеряют ЭДС эталонных растворов, тщательно промывая дистиллированной водой электроды и стакан перед заполнением ячейки каждым эталонным раствором. По полученным данным строят градуировочный график в координатах где с
- концентрация определяемо-
где с
- концентрация определяемо-
го вещества в эталонном растворе. Обычно такой график представляет собой прямую линию.
Затем в электрохимическую ячейку вносят (после промывания ячейки дистиллированной водой) анализируемый раствор и измеряют ЭДС ячейки. По градуировочному графику находят , где - концентрация определяемого вещества в анализируемом растворе.
, где - концентрация определяемого вещества в анализируемом растворе.
. Метод добавок стандарта. В электрохимическую ячейку вносят известный объем V(X) анализируемого раствора с концентрацией и измеряют ЭДС ячейки. Затем в тот же раствор прибавляют точно измеренный небольшой объем стандартного раствора с известной, до-
статочно большой концентрацией определяемого вещества и снова определяют ЭДС ячейки.
Рассчитывают концентрацию определяемого вещества в анализируемом растворе по формуле (17):
(17)
где - разность двух измеренных значений ЭДС;- число электронов, участвующих в электродной реакции.
Применение прямой потенциометрии. Метод применяется для определения концентрации ионов водорода (рН растворов), анионов, ионов металлов (ионометрия).
Большую роль при использовании прямой потенциометрии играет выбор подходящего индикаторного электрода и точное измерение равновесного потенциала.
При определении рН растворов в качестве индикаторных используют электроды, потенциал которых зависит от концентрации ионов водорода: стеклянный, водородный, хингидронный и некоторые другие. Чаще применяют мембранный стеклянный электрод, обратимый по ионам водорода. Потенциал такого стеклянного электрода определяется концентрацией ионов водорода, поэтому ЭДС цепи, включающей стеклянный электрод в качестве индикаторного, описывается при комнатной температуре уравнением:

где постоянная K зависит от материала мембраны, природы электрода сравнения.
Стеклянный электрод позволяет определять рН в интервале рН 0-10 (чаще в диапазоне рН 2-10) и обладает высокой обратимостью и стабильностью в работе.
Хингидронный электрод, часто применявшийся ранее, - окислительно-восстановительный электрод, потенциал которого зависит от концентрации ионов водорода. Он представляет собой платиновую проволоку, погруженную в раствор кислоты (обычно НС1), насыщенный хингидроном - эквимолекулярным соединением хинона с гидрохиноном состава (темно-зеленый порошок, малорастворимый в воде). Схематическое обозначение хингидронного электрода:
На хингидронном электроде протекает окислительно-восстановительная реакция:
Потенциал хингидронного электрода при комнатной температуре описывается формулой:

Хингидронный электрод позволяет измерять рН растворов в интервале рН 0-8,5. При рН < 0 хингидрон гидролитически расщепляется; при рН >8,5 гидрохинон, являющийся слабой кислотой, вступает в реакцию нейтрализации.
Хингидронный электрод нельзя применять в присутствии сильных окислителей и восстановителей.
Мембранные ион-селективные электроды используют в ионометрии в качестве индикаторных для определения различных катионов
И др.) и анионов ,
,
![]() и др.).
и др.).
К достоинствам прямой потенциометрии относятся простота и быстрота проведения измерений. Для измерений требуются небольшие объемы растворов.
Потенциометрическое титрование
Потенциометрическое титрование - способ определения объема титранта, затраченного на титрование определяемого вещества в анализируемом растворе, путем измерения ЭДС (в процессе титрования) с помощью гальванической цепи, составленной из индикаторного электрода и электрода сравнения. При потенциометрическом титровании анализируемый раствор, находящийся в электрохимической ячейке, титруют подходящим титрантом, фиксируя конец титрования по резкому изменению ЭДС измеряемой цепи - потенциала индикаторного электрода, который зависит от концентрации соответствующих ионов и резко изменяется в точке эквивалентности.
Измеряют изменение потенциала индикаторного электрода в процессе титрования в зависимости от объема прибавленного титранта. По полученным данным строят кривую потенциометрического титрования и по этой кривой определяют объем израсходованного титранта в ТЭ.
При потенциометрическом титровании не требуется использование индикаторов, изменяющих окраску вблизи ТЭ.
Электродную пару (электрод сравнения и индикаторный электрод) составляют так, чтобы потенциал индикаторного электрода зависел от концентрации ионов, участвующих или образующихся в реакции, протекающей при титровании. Потенциал электрода сравнения во время титрования должен оставаться постоянным. Оба электрода устанавливают непосредственно в электрохимической ячейке или же помещают в отдельные сосуды с токопроводящими растворами (индикаторный электрод - в анализируемый раствор), которые соединяют электролитическим мостиком, заполненным индифферентным электролитом.
Титрант прибавляют равными порциями, каждый раз измеряя разность потенциалов. В конце титрования (вблизи ТЭ) титрант прибавляют по каплям, также измеряя разность потенциалов после прибавления очередной порции титранта.
Разность потенциалов между электродами измеряют, используя высокоомные потенциометры.
Кривые потенциометрического титрования
Кривая потенциометрического титрования - графическое изображение изменения ЭДС электрохимической ячейки в зависимости от объема прибавленного титранта.
Кривые потенциометрического титрования строят в различных координатах:
Кривые титрования в координатах  , иногда такие кривые называют интегральными кривыми титрования;
, иногда такие кривые называют интегральными кривыми титрования;
Дифференциальные кривые титрования - в координатах
Кривые титрования по методу Грана - в координатах

где- ЭДС потенциометрической ячейки, - объем прибавленно-
- объем прибавленно-
го титранта, - изменение потенциала, соответствующее прибавлению титранта.
На рис. 3-8 приведены схематически различные типы кривых потенциометрического титрования.
По построенным кривым титрования определяют объем титранта
 в ТЭ, как это показано на рис. 3-8. Объем титранта
в ТЭ, как это показано на рис. 3-8. Объем титранта  прибавленного в ТЭ, можно определить
прибавленного в ТЭ, можно определить
не только графически, но и расчетным путем по формуле (18):
где- объем прибавленного титранта, соответствующий последнему измерению до ТЭ;- объем прибавленного титранта, соответствующий первому измерению после ТЭ;


Рис. 3-8.
Типы кривых потенциометрического титрования (Е - измеряемая ЭДС, - объем прибавленного титранта, ![]() - объем титранта, при-
- объем титранта, при-
бавленного в точке эквивалентности): а - кривая титрования в координатах ![]() ; б, в - дифференциальные кривые титрования; г - кривая титрования по методу Грана
; б, в - дифференциальные кривые титрования; г - кривая титрования по методу Грана
В таблице 3-9 в качестве примера (фармакопейного) приведены результаты определений и расчетов при потенциометрическом титровании.
Рассчитаем по формуле (18) величину V
(ТЭ) с использованием данных табл. 3-9. Очевидно, что максимальное значение![]() = 1000. Следовательно,= 5,20 и= 5,30;= 720, .= -450. Отсюда:
= 1000. Следовательно,= 5,20 и= 5,30;= 720, .= -450. Отсюда:
Таблица 3-9. Пример обработки результатов потенциометрического титрования

Применение потенциометрического титрования. Метод - универсальный, его можно применять для индикации конца титрования во всех типах титрования: кислотно-основном, окислительновосстановительном, комплексиметрическом, осадительном, при титровании в неводных средах. В качестве индикаторных используют стеклянный, ртутный, ион-селективные, платиновый, серебряный электроды, а в качестве электродов сравнения - каломельный, хлорсеребряный, стеклянный.
Метод обладает высокой точностью, большой чувствительностью; позволяет проводить титрование в мутных, окрашенных, неводных средах, раздельно определять компоненты смеси в одном анализируемом растворе, например, раздельно определять хлорид- и иодид-ионы при аргентометрическом титровании.
Методами потенциометрического титрования анализируют многие лекарственные вещества, например, аскорбиновую кислоту, сульфамидные препараты, барбитураты, алкалоиды и др.
Задание для самоподготовки к лабораторным занятиям по теме «Потенциометрический анализ»
Цель изучения темы
На основе знания теории потенциометрического анализа и выработки практических умений научиться обоснованно выбирать и практически применять методы прямой потенциометрии и потенциометрического титрования для количественного определения вещества; уметь проводить статистическую оценку результатов потенциометрического анализа.
Целевые задачи
1. Научиться проводить количественное определение содержания фторид-иона в растворе методом прямой потенциометрии с применением фторид-селективного электрода.
2. Научиться проводить количественное определение массовой доли новокаина в препарате методом потенциометрического титрования.
На изучение темы отводятся два лабораторных занятия. На одном занятии студенты выполняют первую лабораторную работу и решают типовые расчетные задачи по основным разделам потенциометрического анализа; на другом занятии студенты выполняют вторую лабораторную работу. Последовательность проведения занятий особого значения не имеет.
Список литературы
1.Учебник. - Книга 2, глава 10. - С. 447-457; 493-507; 510-511.
2.Харитонов Ю.Я. Григорьева В.Ю. Примеры и задачи по аналитической химии.- М.: ГЭОТАР-Медиа, 2007. - С. 214-225; 245-259; 264-271.
3.Лекции по теме: «Потенциометрический анализ».
4.Ефременко О.А. Потенциометрический анализ.- М.: ММА им. И.М. Сеченова, 1998.
К занятию необходимо знать
1. Принцип методов потенциометрического анализа. Уравнение Нернста.
2. Разновидности методов потенциометрического анализа.
3. Схему установки для прямой потенциометрии.
4. Индикаторные электроды и электроды сравнения, применяемые в прямой потенциометрии.
5. Сущность определения концентрации вещества методом прямой потенциометрии с помощью градуировочного графика.
6. Сущность определения содержания фторид-иона в растворе методом прямой потенциометрии с применением фторидселективного электрода.
К занятию необходимо уметь
1. Рассчитывать массу навески для приготовления стандартного раствора вещества.
2. Готовить стандартные растворы методом разбавления.
3. Строить градуировочные графики и использовать их для количественного определения вещества.
Вопросы для самопроверки
1. Какой принцип лежит в основе метода прямой потенциометрии?
3. Какой электрохимический параметр измеряют при определении вещества методом прямой потенциометрии?
4. Приведите схему установки для определения вещества методом прямой потенциометрии.
5. Какие электроды называют индикаторными? Назовите наиболее употребительные индикаторные ион-селективные электроды.
6. Какие электроды называют электродами сравнения? Какой электрод сравнения принят в качестве международного стандарта? Как он устроен? Назовите наиболее часто применяемые электроды сравнения. Как устроены:
а) насыщенный каломельный электрод;
б) насыщенный хлорсеребряный электрод?
7. В чем сущность потенциометрического определения вещества методом градуировочного графика?
8. Назовите диапазон определяемых концентраций и процентную (относительную) погрешность определения вещества методом прямой потенциометрии.
9. Какой принцип лежит в основе определения фторид-иона методом прямой потенциометрии? Перечислите основные этапы анализа.
Лабораторная работа «Определение содержания фторид-иона в растворе с применением фторидселективного электрода»
Цель работы
Научиться применять метод прямой потенциометрии с использованием ион-селективного электрода для количественного определения вещества методом градуировочного графика.
Целевые задачи
1. Приготовление стандартного раствора натрия фторида, концентрация которого точно равна заданной.
2. Приготовление методом разбавления серии стандартных растворов натрия фторида, по составу и ионной силе близких к анализируемому раствору.
3. Измерение электродвижущей силы (ЭДС) гальванического элемента, составленного из индикаторного фторид-селективного электрода и хлорсеребряного электрода сравнения, как функции концентрации фторид-иона.
4. Построение градуировочного графика в координатах: «ЭДС - показатель концентрации фторид-иона».
5. Определение содержания фторид-иона в анализируемом растворе с помощью градуировочного графика.
Материальное обеспечение
Реактивы
1. Натрия фторид, х.ч.
2. Раствор буферный ацетатный, рН ~6.
3. Вода дистиллированная. Лабораторная посуда
1. Колба мерная на 100 мл - 1 шт.
2. Колба мерная на 50 мл - 6 шт.
3. Пипетка мерная на 5 мл - 1 шт.
4. Стакан химический на 200-250 мл - 1 шт.
5. Стакан химический на 50 мл - 2 шт.
6. Бюкс - 1 шт.
7. Воронка - 1 шт.
8. Палочка стек лянная - 1 шт.
9. Промывалка на 250 или 500 мл - 1 шт.
Приборы
2. Электрод индикаторный, фторид-селективный. Перед эксплуатацией фторидный электрод выдерживают в 0,01 моль/л растворе натрия фторида в течение 1-2 ч.
3. Электрод сравнения, вспомогательный лабораторный хлорсеребряный ЭВЛ-IМЗ или аналогичный. Перед эксплуатацией хлорсеребряный электрод наполняют через боковое отверстие концентрированным, но ненасыщенным, примерно 3 моль/л, раствором калия хлорида. При применении насыщенного раствора калия хлорида возможна кристаллизация соли непосредственно вблизи контактной зоны электрода с измеряемым раствором, что препятствует прохождению тока и приводит к невоспроизводимым показаниям измерительного прибора. После заполнения электрода 3 моль/л раствором калия хлорида боковое отверстие закрывают резиновой пробкой, электрод погружают в раствор калия хлорида той же концентрации и выдерживают в этом растворе в течение ~48 ч. В процессе работы пробка из бокового отверстия электрода должна быть удалена. Скорость истечения раствора калия хлорида через электролитический ключ электрода при температуре 20±5 °C составляет 0,3-3,5 мл/сут.
4. Штатив для закрепления двух электродов.
5. Мешалка магнитная.
Прочие материалы
1. Полоски фильтровальной бумаги 3 5 см.
2. Бумага миллиметровая 912 см.
3. Линейка.
Сущность работы
Определение фторид-иона методом прямой потенциометрии основано на измерении электродвижущей силы гальванического элемента, в котором индикаторным электродом служит фторид-селективный электрод, а электродом сравнения - хлорсеребряный или каломельный, как функции концентрации фторид-ионов в растворе.
Чувствительной частью фторидного электрода (рис. 3-9) является мембрана из монокристалла лантана(III) фторида, активированного европием(II).

Рис. 3-9.
Схема устройства фторид-селективного электрода: 1 - мембрана из монокристалла 2 - внутренний полуэлемент (обычно хлорсеребря-
2 - внутренний полуэлемент (обычно хлорсеребря-
ный); 3 - внутренний раствор с постоянной активностью ионов (0,01 моль/л имоль/л); 4 - корпус электрода; 5 - провод для подключения электрода к измерительному прибору
Равновесный потенциал фторидного электрода в соответствии с уравнением Нернста для анион-селективных электродов зависит от активности (концентрации) фторид-иона в растворе:
 (19) или при 25 °C:
(19) или при 25 °C:
(20)
где- стандартный потенциал фторидного электрода, В; -
-
соответственно активность, коэффициент активности, молярная концентрация фторид-иона в растворе.
Первый член правой части уравнения (20)- величина постоянная. Для растворов с примерно одинаковой ионной силой коэффициент активности фторид-иона, а следовательно, и второй член правой части уравнения (20) также является постоянной величиной. Тогда уравнение Нернста можно представить в виде:
Е = const - 0,0591gc (F -) = const + 0,059pF, (21)
где pF = -1gc(F -) - показатель концентрации фторид-иона в растворе.
Таким образом, при постоянной ионной силе
растворов равновесный потенциал фторидного электрода находится в линейной зависимости от показателя концентрации фторид-иона. Существование такой зависимости позволяет проводить определение концентрации фторид-иона с помощью градуировочного графика, который строят в координатах  для серии стандартных растворов натрия фторида, по составу и ионной силе близких анализируемому раствору.
для серии стандартных растворов натрия фторида, по составу и ионной силе близких анализируемому раствору.
Фторидный электрод применяют в диапазоне значений рН 5-9, так как при рН < 5 наблюдается неполная ионизация или образование  а при рН > 9 - взаимодействие материала электрода с гидроксидионом:
а при рН > 9 - взаимодействие материала электрода с гидроксидионом:
Для поддержания постоянного значения рН и создания в стандартных и анализируемых растворах постоянной ионной силы обычно используют буферный раствор (например, ацетатный или цитратный). При анализе растворов со сложным солевым составом буферный раствор служит также для устранения мешающего влияния посторонних катионов путем связывания их в устойчивые ацетатные, цитратные или другие комплексные соединения. С этой же целью в буферный раствор вводят дополнительные комплексообразующие реагенты (например, ЭДТА).
Селективность определения с помощью фторидного электрода очень высокая; мешают только гидроксид-ионы и те немногие катионы, которые образуют с фторид-ионом более устойчивые комплексные соединения, чем с компонентами буферного раствора 
Диапазон определяемых концентраций фторид-иона очень широкий: от 10 -6 до 1 моль/л; при этом процентная погрешность определения составляет ±2%.
Фторид-селективный электрод широко применяется в анализе разнообразных объектов: питьевой воды, фармацевтических препаратов, биологических материалов, при контроле за загрязнением окружающей среды и т.д.
Поскольку в настоящей работе анализируют растворы натрия фторида, не содержащие посторонних ионов, буферный раствор можно не применять. В таком случае следует ожидать небольшого отклонения градуировочного графика от линейной зависимости, так как в стандартных растворах с увеличением концентрации фторид-иона увеличивается ионная сила, и коэффициент активности фторид-иона не сохраняется постоянным.
Порядок выполнения работы
1. (см. приложение 1).
2. Знакомство с назначением, принципом работы и «Инструкцией по эксплуатации универсального иономера ЭВ-74» (или аналогичного прибора) (см. приложения 2, 3).
3.
ВНИМАНИЕ! В данной работе предусмотрено использование иономера типа ЭВ-74. При использовании приборов другого типа необходимо давать дополнительно их описание.
3.1. Собирают гальванический элемент из индикаторного фторидселективного электрода и хлорсеребряного электрода сравнения.
ВНИМАНИЕ! При работе с ион-селективными электродами необходимо соблюдать осторожность, чтобы не повредить рабочей поверхности электрода - мембраны, которая должна быть гладкой, без царапин и отложений.
Перед установкой фторидный электрод энергично встряхивают, как медицинский термометр, держа его в вертикальном положении мембраной вниз. Это делают для того, чтобы удалить невидимые снаружи пузырьки воздуха, которые могут образовываться между поверхностью мембраны и внутренним раствором электрода (см. рис. 3-9) и приводить к нестабильности показаний измерительного прибора.
Фторидный электрод закрепляют в штативе рядом с электродом сравнения.
ВНИМАНИЕ! Держатели, предназначенные для закрепления в штативе электродов, обычно заранее установлены надлежащим образом; не рекомендуется изменять их положение. Для того чтобы закрепить фторидный электрод или поменять раствор в ячейке, следует сначала осторожно убрать из-под ячейки магнитную мешалку.
При закреплении фторидный электрод подводят в лапку штатива снизу так, чтобы его нижний конец оказался на одном уровне с нижним концом электрода сравнения. Электрод подключают к иономеру через гнездо «Изм.», находящееся на задней панели прибора (приложение 3, п. 1.1). Электрод сравнения должен быть подключен к иономеру через гнездо «Всп.».
Электроды многократно промывают дистиллированной водой из промывалки над стаканом вместимостью 200-250 мл, после чего под электроды подводят стакан вместимостью 50 мл с дистиллированной водой, который устанавливают в центре столика магнитной мешалки. Правильно закрепленные электроды не должны касаться стенок и дна
стакана, а также магнитного стержня, применяемого в дальнейшем для перемешивания раствора.
3.2. Иономер включают в сеть под наблюдением преподавателя, руководствуясь инструкцией по эксплуатации прибора (приложение 3, п.п. 1.2-1.7). Дают прибору прогреться в течение 30 мин.
4. Приготовление стандартного 0,1000 моль/л раствора натрия фторида. Рассчитывают с точностью до 0,0001 г массу навески натрия фторида, требуемую для приготовления 100 мл 0,1000 моль/л раствора по формуле:

где с,- соответственно молярная концентрация (моль/л) и объем (л) стандартного раствора натрия фторида;- молярная масса натрия фторида, г/моль.
На аналитических весах с точностью до ±0,0002 г взвешивают сначала чистый и сухой бюкс, а затем в этом бюксе взвешивают навеску х.ч. натрия фторида, масса которого должна быть точно вычисленной.
Взятую навеску количественно переносят в мерную колбу вместимостью 100 мл через сухую воронку, смывая частицы соли со стенок бюкса и воронки ацетатным буферным раствором (рН ~6). Раствор из бюкса сливают в колбу по стеклянной палочке, прислонив ее к краю бюкса. Добиваются полного растворения соли, после чего буферным раствором доводят объем раствора до метки колбы. Содержимое колбы перемешивают.
5. Приготовление серии стандартных растворов натрия фторида с постоянной ионной силой. Серию стандартных растворов с концентрацией фторид-иона, равной 10 -2 , 10 -3 , 10 -4 , 10 -5 и 10 -6 моль/л, готовят в мерных колбах вместимостью 50 мл из стандартного 0,1000 моль/л раствора натрия фторида путем последовательного разбавления буферным раствором.
Так, для приготовления 10 -2 моль/л раствора в мерную колбу на 50 мл помещают пипеткой 5 мл 0,1000 моль/л раствора натрия фторида, предварительно ополоснув пипетку небольшим количеством этого раствора 2-3 раза, буферным раствором доводят объем раствора до метки, содержимое колбы перемешивают. Таким же способом из 10 -2 моль/л раствора готовят 10 -3 моль/л раствор и т.д. вплоть до 10 -6 моль/л раствора натрия фторида.
6. Измерение электродвижущей силы гальванического элемента как функции концентрации фторид-иона. В стакан вместимостью 50 мл последовательно помещают приготовленные стандартные растворы на-
трия фторида, начиная с самого разбавленного, предварительно ополоснув стакан измеряемым раствором 2-3 раза. Осторожно осушают поверхность фторидного и хлорсеребряного электродов фильтровальной бумагой, после чего электроды погружают в измеряемый раствор, опускают магнитный стержень и устанавливают ячейку в центре столика магнитной мешалки. Если на то будет указание преподавателя, открывают боковое отверстие хлорсеребряного электрода, удалив из него резиновую пробку. Включают магнитную мешалку и измеряют ЭДС элемента (положительный потенциал фторидного электрода) с помощью иономера ЭВ-74 на узком диапазоне измерений - 14 так, как указано в Приложении 3, п.п. 2.1-2.5. Результаты измерений заносят в табл. 3-10.
Таблица 3-10. Результаты измерения электродвижущей силы гальванического элемента как функции концентрации фторид-иона
7. Построение градуировочного графика. По данным табл. 3-10 на миллиметровой бумаге строят градуировочный график, откладывая по оси абсцисс показатель концентрации фторид-иона а по оси ординат - ЭДС элемента в милливольтах (Е, мВ). Если выполняется зависимость (21), то получается прямая, тангенс угла наклона которой к оси абсцисс составляет 59±2 мВ (при 25 °C). График подклеивают в лабораторный журнал.
8. Определение содержания фторид-иона в анализируемом растворе с помощью градуировочного графика. Анализируемый раствор, содержащий фторид-ион, получают от преподавателя в мерной колбе на 50 мл. Объем раствора доводят до метки ацетатным буферным раствором. Содержимое колбы перемешивают и в полученном растворе измеряют ЭДС элемента, составленного из фторидного и хлорсеребряного электродов.
По окончании измерений закрывают отверстие хлорсеребряного электрода резиновой пробкой и выключают прибор, как указано в Приложении 3, п. 2.6.
По градуировочному графику находят показатель концентрации фторид-иона, соответствующий ЭДС элемента в анализируемом растворе, затем определяют молярную концентрацию и рассчитывают содержание фторид-иона в растворе по формуле:

где  - титр фторид-иона в анализируемом растворе, г/мл; - моляр-
- титр фторид-иона в анализируемом растворе, г/мл; - моляр-
ная концентрация фторид-иона, найденная с помощью градуировочного графика, моль/л;  - молярная масса фторид-иона, г/моль.
- молярная масса фторид-иона, г/моль.
Расчет титра проводят с точностью до трех значащих цифр.
9. Определение содержания фторид-иона в анализируемом растворе по уравнению градуировочного графика. Значение рF для анализируемого раствора можно найти по уравнению градуировочного графика, что представляется более точным, чем с помощью градуировочного графика. Это уравнение имеет вид:

где ![]() цепи с испытуемым раствором
цепи с испытуемым раствором![]() ;
;![]() цепи при = 0 -
цепи при = 0 -
отрезок, отсекаемый прямой по оси ординат![]() ;
;![]() - тангенс угла
- тангенс угла
наклона прямой к оси абсцисс:

где n - количество эталонных растворов. Таким образом:

Определив по графикуи рассчитав рассчитывают
рассчитывают
по формуле:

Затем определяют молярную концентрацию и рассчитывают содержание фторид-иона в растворе по формуле, указанной выше.
Контрольные вопросы
1. Назовите составные части гальванического элемента, служащего для определения концентрации (активности) фторид-иона в растворе методом прямой потенциометрии.
2. Какая математическая зависимость лежит в основе определения концентрации (активности) фторид-иона в растворе методом прямой потенциометрии?
3. Опишите устройство фторид-селективного электрода. От каких факторов зависит его потенциал?
4. Почему при определении концентрации фторид-иона методом прямой потенциометрии в анализируемом и стандартных растворах необходимо создавать одинаковую ионную силу?
5. Какой диапазон значений рН является оптимальным для определения фторид-иона с помощью фторид-селективного электрода?
6. Каким образом при определении фторид-иона в растворах со сложным солевым составом поддерживают оптимальное значение рН и постоянную ионную силу?
7. Какие ионы мешают определению фторид-иона в растворе с помощью фторид-селективного электрода? Как устраняют их мешающее влияние?
8. Перечислите основные этапы определения концентрации фторид-иона в растворе потенциометрическим методом с применением градуировочного графика.
9. В каких координатах строят градуировочный график при определении концентрации фторид-иона методом прямой потенциометрии?
10. Чему должен быть равен угловой коэффициент (тангенс угла наклона) градуировочного графика, построенного в координатах , для стандартных растворов натрия фторида с одинаковой ионной силой при 25 °C?
, для стандартных растворов натрия фторида с одинаковой ионной силой при 25 °C?
11. Как рассчитать концентрацию фторид-иона в растворе с использованием данных градуировочного графика, построенного в координатах , если известна ЭДС элемента в анализируемом растворе?
, если известна ЭДС элемента в анализируемом растворе?
12. Как приготовить из кристаллического вещества натрия фторида стандартный раствор с концентрацией, точно равной заданной, например 0,1000 моль/л?
13. Как приготовить стандартный раствор натрия фторида из более концентрированного раствора?
14. Назовите диапазон определяемых концентраций и процентную погрешность определения фторид-иона с помощью фторидселективного электрода методом градуировочного графика.
15. Назовите области применения фторид-селективного электрода.
Занятие 2. Потенциометрическое титрование
К занятию необходимо знать
1. Принцип методов потенциометрического анализа. Уравнение Нернста. Разновидности методов потенциометрического анализа.
2. Принципиальную схему установки для потенциометрического титрования.
3. Индикаторные электроды, применяемые в потенциометрическом титровании в зависимости от типа реакции титрования; электроды сравнения.
4. Способы индикации точки эквивалентности в потенциометрическом титровании.
5. Преимущества потенциометрического титрования перед титриметрическим анализом с визуальной индикацией точки эквивалентности.
6. Сущность определения новокаина методом потенциометрического титрования.
К занятию необходимо уметь
1. Готовить анализируемый раствор растворением навески испытуемого образца с точно известной массой.
2. Рассчитывать массовую долю вещества в анализируемом образце на основе результатов титрования.
3. Писать уравнение реакции, протекающей при титровании.
Вопросы для самопроверки
1. Какой принцип лежит в основе метода потенциометрического титрования?
2. Каким уравнением выражается зависимость электродного потенциала от концентрации (активности) потенциалопределяющих компонентов в растворе?
3. Какой электрохимический параметр измеряют при определении вещества методом потенциометрического титрования?
4. Дайте определение понятиям «индикаторный электрод», «электрод сравнения».
5. В чем причина резкого изменения электродвижущей силы гальванического элемента (потенциала индикаторного электрода) в титруемом растворе вблизи точки эквивалентности?
6. Назовите известные способы определения точки эквивалентности на основе данных потенциометрического титрования.
7. Для каких типов химических реакций можно использовать метод потенциометрического титрования? Какие электроды применяются при этом?
8. В чем преимущество потенциометрического титрования перед титриметрическим анализом с визуальной индикацией точки эквивалентности?
9. Назовите диапазон определяемых концентраций и процентную (относительную) погрешность определения вещества методом потенциометрического титрования.
10. Какая химическая реакция лежит в основе определения вещества, содержащего первичную ароматическую аминогруппу, методом нитритометрического титрования? Каковы условия ее проведения? Применяемые индикаторы?
11. Какой принцип лежит в основе определения новокаина методом потенциометрического титрования? Перечислите основные этапы анализа.
Лабораторная работа «Определение массовой доли новокаина в препарате»
Цель работы
Научиться применять метод потенциометрического титрования для количественного определения вещества.
Целевые задачи
1. Ориентировочное потенциометрическое титрование новокаина раствором натрия нитрита.
2. Точное потенциометрическое титрование новокаина раствором натрия нитрита.
3. Нахождение конечной точки потенциометрического титрования.
4. Расчет массовой доли новокаина в препарате.
Материальное обеспечение
Реактивы
1. Натрия нитрит, стандартный ~0,1 моль/л раствор.
2. Новокаин, порошок.
3. Калия бромид, порошок.
4. Кислота соляная концентрированная (= 1,17 г/мл).
5. Вода дистиллированная. Лабораторная посуда
1. Колба мерная на 100 мл.
2. Колба мерная на 20 мл.
3. Бюретка на 25 мл.
4. Цилиндр мерный на 20 мл.
5. Цилиндр мерный на 100 мл.
6. Стакан для титрования на 150 мл.
7. Бюкс.
8. Воронка.
9. Промывалка на 250 или 500 мл.
Приборы
1. Иономер универсальный ЭВ-74 или аналогичный.
2. Электрод индикаторный платиновый ЭТПЛ-01 М или аналогичный.
3. Электрод сравнения, вспомогательный лабораторный хлорсеребряный ЭВЛ-1МЗ или аналогичный.
Подготовка хлорсеребряного электрода к эксплуатации - см. выше, предыдущую лабораторную работу.
4. Штатив для закрепления двух электродов и бюретки.
5. Мешалка магнитная.
6. Весы аналитические с разновесом.
7. Весы технохимические с разновесом.
Прочие материалы: см. «Материальное обеспечение» в предыдущей работе.
Сущность работы
Потенциометрическое титрование основано на индикации точки эквивалентности по резкому изменению (скачку) потенциала индикаторного электрода в процессе титрования.
Для определения новокаина - вещества, содержащего первичную ароматическую аминогруппу, - применяют метод нитритометрического титрования, согласно которому новокаин титруют стандартным 0,1 моль/л раствором натрия нитрита в солянокислой среде в присутствии калия бромида (ускоряет протекание реакции) при температуре не выше 18-20 °C. В таких условиях реакция титрования протекает количественно и достаточно быстро:

За ходом реакции диазотирования наблюдают с помощью индикаторного платинового электрода, который в паре с подходящим электродом сравнения (хлорсеребряным или каломельным) погружают в титруемый раствор, и измеряют электродвижущую силу  элемента в зави-
элемента в зави-
симости от объема прибавленного титранта
Потенциал индикаторного электрода согласно уравнению Нернста зависит от концентрации (активности) веществ, участвующих в реакции титрования. Вблизи точки эквивалентности (ТЭ) концентрация потенциалопределяющих веществ резко изменяется, что сопровождается резким изменением (скачком) потенциала индикаторного электрода. ЭДС элемента определяется разностью потенциалов между индикаторным электродом и электродом сравнения. Поскольку потенциал электрода сравнения сохраняется постоянным, скачок потенциала индикаторного электрода вызывает резкое изменение ЭДС элемента, что указывает на достижение ТЭ. Для большей точности определения ТЭ титрант в конце титрования прибавляют по каплям.
Графические способы, обычно применяемые для нахождения ТЭ, в данном случае применять вряд ли целесообразно, так как кривая титрования, построенная в координатах , асимметрична относительно ТЭ; установить ТЭ с достаточно высокой точностью довольно сложно.
, асимметрична относительно ТЭ; установить ТЭ с достаточно высокой точностью довольно сложно.
Процентная погрешность определения новокаина в препарате методом потенциометрического титрования не превышает 0,5%.
Аналогично определению новокаина методом потенциометрического титрования можно определять многие другие органические соединения и лекарственные препараты, содержащие первичную ароматическую аминогруппу, например, сульфацил, норсульфазол, производные n-аминобензойной кислоты и др.
Примечание. Реакция диазотирования протекает медленно. На скорость ее протекания влияют различные факторы. Увеличение кислотности приводит к уменьшению скорости реакции, поэтому при титровании стараются избегать большого избытка соляной кислоты. Для ускорения реакции в реакционную смесь вводят калия бромид. Температура оказывает обычное влияние
на скорость реакции: повышение температуры на 10 °C приводит к увеличению скорости примерно в 2 раза. Однако титрование, как правило, проводят при температуре не выше 18-20 °C, а во многих случаях еще ниже, при охлаждении реакционной смеси до 0-10 °C, так как образующиеся в результате реакции диазосоединения неустойчивы и при более высокой температуре разлагаются.
Титрование с применением реакции диазотирования проводят медленно: сначала со скоростью 1-2 мл/мин, а в конце титрования - 0,05 мл/мин.
Порядок выполнения работы
ВНИМАНИЕ! В данной работе предусмотрено применение универсального иономера ЭВ-74. При использовании приборов другого типа необходимо дополнительно давать их описание в лабораторных методических указаниях.
1. Знакомство с «Инструкцией по технике безопасности при работе с электроприборами» (см. Приложение 1).
2. Знакомство с назначением, принципом работы и «Инструкцией по эксплуатации универсального иономера ЭВ-74» (см. Приложения 2, 3) или аналогичного прибора.
3. Подготовка иономера к измерениям.
3.1. Собирают гальванический элемент из индикаторного платинового электрода и хлорсеребряного электрода сравнения.
Платиновый электрод закрепляют в штативе рядом с электродом сравнения.
ВНИМАНИЕ! Держатели, предназначенные для закрепления в штативе электродов и бюретки, обычно заранее установлены надлежащем образом. Их положение изменять не рекомендуется. Для того чтобы закрепить платиновый электрод или заменить раствор в ячейке, следует сначала осторожно убрать из-под ячейки магнитную мешалку.
Для закрепления платиновый электрод подводят в лапку штатива снизу так, чтобы его нижний конец оказался несколько выше (примерно на 0,5 см) нижнего конца электрода сравнения. Индикаторный электрод подключают к иономеру через гнездо «Изм.», находящееся на задней панели прибора (см. Приложение 3, п. 1.1). Электрод сравнения должен быть подключен к иономеру через гнездо «Всп.».
Электроды многократно промывают дистиллированной водой из промывалки над стаканом на 200-250 мл, после чего под электроды подводят стакан на 150 мл с дистиллированной водой, который устанавливают в центре столика магнитной мешалки. Правильно закрепленные электроды не должны касаться стенок и дна стакана, а также магнитного стержня, применяемого в дальнейшем для перемешивания раствора.
3.2. Иономер включают в сеть под наблюдением преподавателя, руководствуясь инструкцией по эксплуатации прибора (Приложение 3, п.п. 1.2-1.7). Дают прибору прогреться в течение 30 мин.
4. Приготовление анализируемого раствора новокаина. Готовят примерно 0,05 моль/л раствор новокаина в 2 моль/л растворе соляной кислоты. Для этого около 0,9 г препарата (навеску взвешивают в бюксе на аналитических весах с точностью до ±0,0002 г) помещают в мерную колбу на 100 мл, добавляют 20-30 мл дистиллированной воды, 16,6 мл концентрированного раствора соляной кислоты (= 1,17 г/мл). Смесь перемешивают до полного растворения препарата, доводят объем раствора до метки дистиллированной водой, содержимое колбы перемешивают.
5. Ориентировочное титрование. В стакан вместимостью 150 мл пипеткой помещают 20 мл анализируемого раствора новокаина, прибавляют 60 мл дистиллированной воды с помощью цилиндра и около 2 г калия бромида. Электроды - индикаторный платиновый и вспомогательный хлорсеребряный - погружают в титруемый раствор, опускают магнитный стержень и устанавливают ячейку в центре столика магнитной мешалки. Если на то будет указание преподавателя, открывают боковое отверстие хлорсеребряного электрода, удалив из него резиновую пробку. Бюретку на 25 мл наполняют стандартным 0,1 моль/л раствором натрия нитрита и закрепляют в штативе так, чтобы нижний конец бюретки был опущен в стакан на 1-2 см ниже его края. Включают магнитную мешалку. Перемешивание не прекращают в течение всего процесса титрования.
Прибор включают в режим милливольтметра для измерения положительных потенциалов (+мВ). При ориентировочном титровании измерение ЭДС системы производят на широком диапазоне (-119) так, как указано в Приложении 3, п.п. 2.1-2.5, раствор титранта прибавляют порциями по 1 мл, каждый раз измеряя ЭДС системы после того, как показание прибора примет установившееся значение.
Наблюдают резкое изменение ЭДС (скачок титрования), а затем прибавляют еще 5-7 мл титранта порциями по 1 мл и убеждаются в незначительном изменении измеряемой величины. По окончании титрования выключают магнитную мешалку. Результаты измерений заносят в табл. 3-11.
На основании результатов ориентировочного титрования устанавливают объем титранта, после добавления которого наблюдается скачок титрования. Этот объем считают близким к объему, соответствующему конечной точке титрования (КТТ).
В приведенном в табл. 3-11 примере объем титранта, затраченный на ориентировочное титрование, составляет 11 мл.
Таблица 3-11. Ориентировочное титрование (пример)

По результатам ориентировочного титрования строят кривую титрования в координатахОтмечают асимметричный характер кривой, затрудняющий определение КТТ графическим способом с надлежащей точностью.
6. Точное титрование. В чистый стакан на 150 мл помещают новую порцию анализируемого раствора новокаина, дистиллированную воду, калия бромид в тех же количествах, что и при ориентировочном титровании. В раствор погружают электроды, предварительно промытые дистиллированной водой, опускают магнитный стержень и включают магнитную мешалку. При точном титровании измерение ЭДС проводят на узком диапазоне (49) так, как указано в приложении 3, п. 2.5.
Сначала к титруемому раствору со скоростью 1 мл/мин прибавляют такой объем титранта, который должен быть на 1 мл меньше объема, затраченного на ориентировочное титрование, после чего измеряют ЭДС элемента. В приведенном примере объем прибавленного титранта составляет: 11 - 1 = 10 мл.
Затем титрант прибавляют порциями по 2 капли, каждый раз измеряя ЭДС после того, как показание прибора примет установившееся значение. Наблюдают резкое изменение ЭДС (скачок титрования), а затем продолжают титрование порциями по 2 капли и убеждаются в уменьшении и небольшом изменении По окончании титрования отмечают общий объем добавленного титранта с точностью до сотых долей миллилитра.
Выключают магнитную мешалку. Результаты титрования заносят в табл. 3-12.
Точное титрование проводят не менее трех раз. По окончании измерений закрывают отверстие хлорсеребряного электрода резиновой пробкой и выключают прибор, как указано в Приложении 3, п. 2.6.
7.
Расчет результата анализа.
На основании данных точного титрования вычисляют сначала объем одной капли а затем объем титранта, соответствующий по формулам:
по формулам:

где- объем титранта, после прибавления которого титрование продолжают по каплям, мл;- объем титранта в конце титрования, мл; n - общее число добавленных капель титранта;- число капель титранта, добавленных до появления скачка титрования;- число капель, составляющих порцию раствора титранта, вызвавшую скачок титрования.
Таблица 3-12. Точное титрование (пример)

Пример. Расчет по данным табл. 3-12.

Объем титранта , затраченный на титрование, определяют для каждого i-го титрования.
Массовую долю (в процентах) новокаина в препарате рассчи-
рассчи-
тывают с точностью до сотых долей процента по формуле:

где с - молярная концентрация титранта: стандартного раствора натрия нитрита, моль/л; - объем титранта, затраченный на i-е точное титрование, мл;
Объем аликвотной доли раствора новокаина, мл; - общий объем анализируемого раствора новокаина, мл; M - молярная масса новокаина, равная 272,78 г/моль; m - масса навески препарата, содержащего новокаин, г.
Полученные значения массовой доли новокаина в препарате обрабатывают методом математической статистики, представляя результат анализа в виде доверительного интервала для доверительной вероятности 0,95.
Контрольные вопросы
1. В чем состоит принцип определения новокаина методом потенциометрического титрования?
2. Какая химическая реакция лежит в основе определения новокаина методом потенциометрического титрования?
3. С помощью каких электродов можно следить за ходом реакции диазотирования в процессе титрования новокаина раствором натрия нитрита?
4. Чем вызван скачок ЭДС (скачок потенциала индикаторного электрода) в области точки эквивалентности при титровании новокаина раствором натрия нитрита?
5. В каких условиях реакция диазотирования (с участием новокаина) протекает количественно и достаточно быстро?
6. С какой скоростью проводят потенциометрическое титрование новокаина раствором натрия нитрита?
7. Какой вид имеет кривая титрования новокаина раствором натрия нитрита, построенная в координатах «ЭДС - объем титранта»?
8. Целесообразно ли применять графические способы определения точки эквивалентности при потенциометрическом титровании новокаина?
10. Чему равна процентная (относительная) погрешность определения новокаина в препарате методом потенциометрического титрования?
11. Какие преимущества имеет потенциометрический способ индикации точки эквивалентности по сравнению с визуальным при определении новокаина методом нитритометрического титрования?
12. Какие вещества можно определять методом потенциометрического титрования по аналогии с определением новокаина?
Приложение 1
Инструкция по технике безопасности при работе с электроприборами
Работать с незаземленными приборами;
Оставлять включенный прибор без присмотра;
Перемещать включенный прибор;
Работать вблизи открытых токонесущих частей прибора;
Включать и выключать прибор влажными руками.
2. В случае перерыва в подаче электроэнергии немедленно выключить прибор.
3. В случае загорания проводов или электроприбора необходимо немедленно их обесточить и гасить огонь с помощью сухого огнетушителя, покрывала из асбеста, песком, но не водой.
Приложение 2
Назначение и принцип работы универсального иономера ЭВ-74
1. Назначение прибора
Универсальный иономер ЭВ-74 предназначен для определения в комплекте с ионселективными электродами активности (показателя активности - рХ) одно- и двухзарядных ионов (например, ,
, ![]() и др.), а также для измерения окислительно-восстановительных потенциалов (электродвижущей силы) -соответствующих электродных систем в водных растворах электролитов.
и др.), а также для измерения окислительно-восстановительных потенциалов (электродвижущей силы) -соответствующих электродных систем в водных растворах электролитов.
Иономер можно использовать также в качестве высокоомного милливольтметра.
2. Принцип работы прибора
Работа иономера основана на преобразовании электродвижущей силыэлектродной системы в постоянный ток, пропорциональный измеряемой величине. Преобразование осуществляется с помощью высокоомного преобразователя автокомпенсационного типа.
Электродвижущая сила электродной системы сравнивается с противоположным по знаку падением напряжения на прецизионном сопротивлении R, через которое протекает ток усилителя На вход усилителя подается напряжение:
При достаточно большом коэффициенте усиления напряжение мало отличается от электродвижущей силыи благодаря этому ток, протекающий через электроды в процессе измерения, весьма мал, а ток , протекающий через сопротивление R, пропорционален электродвижущей силе электродной системы:

Измерив ток с помощью микроамперметра А, можно определить а также в исследуемом растворе.
Приложение 3
Инструкция по эксплуатации универсального иономера ЭВ-74 для измерения окислительно-восстановительных потенциалов (ЭДС) электродных систем
Измерения могут проводиться как в милливольтах, так и в единицах рХ по шкале прибора. При измерении ЭДС поправка на температуру испытуемого раствора не вводится.
1. Подготовка иономера ЭВ-74 к измерениям.
1.1. Выбирают необходимые электроды и закрепляют их в штативе. Индикаторный электрод подключают к гнезду «Изм.» непосредственно или с помощью переходного штекера, а электрод сравнения - к гнезду «Всп.» на задней панели прибора. Электроды промывают и погружают в стакан с дистиллированной водой.
1.2. Проверяют наличие заземления корпуса прибора.
1.3. Устанавливают механический ноль показывающего прибора, для чего, поворачивая отверткой корректор нуля, устанавливают стрелку на нулевую (начальную) отметку шкалы.
1.4. Нажимают нижнюю кнопку «t°» выбора рода работы и верхнюю кнопку «-119» выбора диапазона измерения.
1.5. Подключают прибор к сети 220 В с помощью шнура.
1.6. Включают прибор с помощью тумблера «Сеть». При подаче напряжения загорается глазок индикации включения.
1.7. Прибор прогревается в течение 30 мин.
2. Измерение окислительно-восстановительных потенциалов (ЭДС) электродных систем.
2.1. Электроды погружают в стакан с испытуемым раствором, предварительно удалив с поверхности электродов избыток дистиллированной воды фильтровальной бумагой.
2.2. Включают магнитную мешалку.
2.3. Нажимают кнопку ![]() и кнопку выбранного диапазона измерения.
и кнопку выбранного диапазона измерения.
2.4. Оставляют отжатой кнопку «анион | катион; +|-», если измеряют положительные потенциалы, и нажимают при измерении отрицательных потенциалов.
2.5. Дают установиться показаниям прибора и проводят отсчет значения потенциала в милливольтах по соответствующей шкале показывающего прибора, умножая показание прибора на 100:
При измерении на широком диапазоне «-119» отсчет проводят по нижней шкале с оцифровкой от -1 до 19;
При измерении на узком диапазоне «-14» отсчет проводят по верхней шкале с оцифровкой от -1 до 4;
При измерении на одном из узких диапазонов «49», «914», «1419» отсчет проводят по верхней шкале с оцифровкой от 0 до 5, причем показание прибора суммируют со значением нижнего предела выбранного диапазона.
Пример. Переключатель диапазонов установлен в положение «49», а стрелка прибора установилась на значении 3,25. В этом случае измеряемая величина равна: (4 + 3,25) . 100=725 мВ.
2.6. По окончании измерений нажимают на кнопку «t°» и «-119», выключают прибор с помощью тумблера «Сеть» и отключают прибор и магнитную мешалку от сети. Электроды и стержень магнитной мешалки промывают дистиллированной водой и сдают лаборанту.
Занятие 3. Кулонометрический анализ Принцип метода
Кулонометрический анализ (кулонометрия) основан на использовании зависимости между массой m вещества, прореагировавшего при электролизе в электрохимической ячейке, и количеством электричества Q, прошедшего через электрохимическую ячейку при электролизе только этого вещества. В соответствии с объединенным законом электролиза М. Фарадея масса m (в граммах) связана с количеством электричества Q (в кулонах) соотношением:
 (1)
(1)
где M - молярная масса вещества, прореагировавшего при электролизе, г/моль; n - число электронов, участвующих в электродной реакции; F = 96 487 Кл/моль - число Фарадея.
Количество электричества(в кулонах), прошедшее при электролизе через электрохимическую ячейку, равно произведению электрического тока(в амперах) на время электролиза(в секундах):
 (2)
(2)
Если измерено количество электричества то согласно (1) можно рассчитать массу m. Это справедливо в том случае, когда все количество электричества прошедшее при электролизе через электрохимическую ячейку, израсходовано только на электролиз данного вещества; побоч-
ные процессы должны быть исключены. Другими словами, выход (эффективность) по току должен быть равен 100%.
Поскольку в соответствии с объединенным законом электролиза М. Фарадея (1) для определения массы m (г) прореагировавшего при электролизе вещества необходимо измерить количество электричества Q, затраченное на электрохимическое превращение определяемого вещества, в кулонах, то метод и назван кулонометрией. Главная задача кулонометрических измерений - как можно более точно определить количество электричества Q.
Кулонометрический анализ проводят либо в амперостатическом (гальваностатическом) режиме, т.е. при постоянном электрическом токе i = const, либо при контролируемом постоянном потенциале рабочего электрода (потенциостатическая кулонометрия), когда электрический ток изменяется (уменьшается) в процессе электролиза.
В первом случае для определения количества электричества Q достаточно как можно более точно измерить время электролиза, постоянный ток и рассчитать величину Q по формуле (2). Во втором случае величину Q определяют либо расчетным способом, либо с помощью химических кулонометров.
Различают прямую и косвенную кулонометрию (кулонометрическое титрование).
Прямая кулонометрия
Сущность метода
Прямую кулонометрию при постоянном токе применяют редко. Чаще используют кулонометрию при контролируемом постоянном потенциале рабочего электрода или прямую потенциостатическую кулонометрию.
В прямой потенциостатической кулонометрии электролизу подвергают непосредственно определяемое вещество. Измеряют количество электричества, затраченное на электролиз этого вещества, и по уравнению (1) рассчитывают массу m определяемого вещества.
В процессе электролиза потенциал рабочего электрода поддерживают постоянным,  для чего обычно используют приборы - потенциостаты.
для чего обычно используют приборы - потенциостаты.
Постоянное значение потенциала E выбирают предварительно на основании рассмотрения вольтамперной (поляризационной) кривой, построенной в координатах «ток i - потенциал Е», полученной в тех же условиях, в которых будет проводиться электролиз. Обычно выбирают
значение потенциала Е, соответствующее области предельного тока для определяемого вещества и несколько превышающее его потенциал полуволны(на ~0,05-0,2 B). При этом значении потенциала фоновый электролит не должен подвергаться электролизу.
В качестве рабочего электрода чаще всего применяют платиновый электрод, на котором происходит электрохимическое восстановление или окисление определяемого вещества. Кроме рабочего электрода электрохимическая ячейка включает 1 или 2 других электрода - электрод сравнения, например, хлорсеребряный, и вспомогательный электрод, например, из стали.
По мере протекания процесса электролиза при постоянном потенциале электрический ток в ячейке уменьшается, так как понижается концентрация электроактивного вещества, участвующего в электродной реакции. При этом электрический ток уменьшается со временем по экспоненциальному закону от начального значения в момент времени до значения в момент времени
 (3)
(3)
где коэффициентзависит от природы реакции, геометрии электрохимической ячейки, площади рабочего электрода, коэффициента диффузии определяемого вещества, скорости перемешивания раствора и его объема.
График функции (3) схематически показан на рис. 3-10.

Рис. 3-10. Изменение токасо временемв прямой потенциостатической кулонометрии
Выход по току будет количественным, когда ток уменьшится до нуля, т.е. при бесконечно большом времени . На практике электролиз
определяемого вещества считают количественным, когда ток достигнет очень малой величины, не превышающей ~0,1% значения При этом ошибка определения составляет около ~0,1%.
Поскольку количество электричества определяется как произведение тока на время электролиза, очевидно, что общее количество электричества Q, затраченное на электролиз определяемого вещества, равно:
 (4)
(4)
т.е. определяется площадью, ограниченной осями координат и экспонентой на рис. 3-10.
Для нахождения массы m прореагировавшего вещества требуется согласно (1) измерить или рассчитать количество электричества Q.
Способы определения количества электричества, прошедшего через раствор, в прямой потенциостатической кулонометрии
Величину Q можно определить расчетными способами либо с помощью химического кулонометра.
. Расчет величины Q по площади под кривой зависимости i от Измеряют площадь, ограниченную осями координат и экспонентой (3) (см. рис. 3-10). Если ток i выражен в амперах, а время - в секундах, то измеренная площадь равна количеству электричества Q в кулонах.
Для определения Q без заметной ошибки способ требует практически полного завершения процесса электролиза, т.е. длительного времени. На практике измеряют площадь при значении т, соответствующем i = 0,001(0,1% от.
. Расчет величины Q на основе зависимости от В соответствии с (3) и (4) имеем:

поскольку:
Таким образом,  и для определения величины Q
необходимо
и для определения величины Q
необходимо
найти значения
Согласно (3) . После логарифмирования этого уравнения по-
. После логарифмирования этого уравнения по-
лучаем линейную зависимость от
 (5)
(5)
Если измерить несколько значенийв различные моменты времени(например, воспользовавшись кривой типа представленной на рис. 3-10 или непосредственно опытным путем), можно построить график функции (5), схематически показанный на рис. 3-11 и представляющий собой прямую линию.
Отрезок, отсекаемый прямой линией на оси ординат, равена тангенс угла наклона прямой к оси абсцисс равен:
Зная значения а следовательно,можно рассчитать величи-
а следовательно,можно рассчитать величи-
ну , а затем и массу m
по формуле (1).
, а затем и массу m
по формуле (1).

Рис. 3-11. Зависимостьот времени электролизав прямой потенциостатической кулонометрии
. Определение величины Q с помощью химического кулонометра. При этом способе в электрическую цепь кулонометрической установки включают химический кулонометр последовательно с электрохимической ячейкой, в которой проводят электролиз определяемого вещества. Количество электричества Q, проходящее через последовательно соединенные кулонометр и электрохимическую ячейку, одинаково. Конструкция кулонометра позволяет экспериментально определить величину Q.
Чаще всего применяют серебряный, медный и газовые кулонометры, реже некоторые другие. Использование серебряного и медного кулонометров основано на электрогравиметрическом определении массы серебра или меди, осаждающейся на платиновом катоде при электролизе.
Зная массу металла, выделившегося на катоде в кулонометре, можно по уравнению (1) рассчитать количество электричества Q.
Кулонометры, особенно серебряный и медный, позволяют определять количество электричества Q с высокой точностью, однако работа с ними довольно трудоемка и продолжительна.
В кулонометрии применяют также электронные интеграторы, позволяющие регистрировать количество электричества Q, затраченное на электролиз, по показаниям соответствующего прибора.
Применение прямой кулонометрии
Метод обладает высокими селективностью, чувствительностью (до 10 -8 -10 -9 г или до ~10 -5 моль/л), воспроизводимостью (до ~1-2%), позволяет определять содержание микропримесей. К недостаткам метода относится большая трудоемкость и длительность проведения анализа, необходимость наличия дорогостоящей аппаратуры.
Прямую кулонометрию можно применять для определения ионов металлов, органических нитро- и галогенпроизводных, хлорид-, бромид-, иодид-, тиоцианат-анионов, ионов металлов в низших степенях окисления при переводе их в более высокие состояния окисления, например:
И т.д.
В фармацевтическом анализе прямую кулонометрию применяют для определения аскорбиновой и пикриновой кислот, новокаина, оксихинолина и в некоторых других случаях.
Прямая кулонометрия довольно трудоемка и продолжительна. Кроме того, в ряде случаев начинают заметно протекать побочные процессы еще до завершения основной электрохимической реакции, что снижает выход по току и может привести к значительным ошибкам анализа. Именно поэтому чаще применяют косвенную кулонометрию - кулонометрическое титрование.
Кулонометрическое титрование
Сущность метода
При кулонометрическом титровании определяемое вещество X, находящееся в растворе в электрохимической ячейке, реагирует с титрантом T - веществом, непрерывно образующимся (генерируемым) на генераторном электроде при электролизе вспомогательного вещества, также присутствующего в растворе. Окончание титрования - момент, когда все определяемое вещество X полностью прореагирует с генерируемым титрантом T, фиксируют либо визуально индикаторным мето-
дом, вводя в раствор соответствующий индикатор, меняющий окраску вблизи ТЭ, либо с помощью инструментальных методов - потенциометрически, амперометрически, фотометрически.
Таким образом, при кулонометрическом титровании титрант не прибавляется из бюретки в титруемый раствор. Роль титранта играет вещество T, непрерывно генерируемое при электродной реакции на генераторном электроде. Очевидно, имеется аналогия между обычным титрованием, когда титрант вводится извне в титруемый раствор и по мере его прибавления реагирует с определяемым веществом, и генерацией вещества T, которое по мере своего образования также реагирует с определяемым веществом, поэтому рассматриваемый метод и получил название «кулонометрическое титрование».
Кулонометрическое титрование проводят в амперостатическом (гальваностатическом) или в потенциостатическом режиме. Чаще кулонометрическое титрование проводят в амперостатическом режиме, поддерживая электрический ток постоянным в течение всего времени электролиза.
Вместо объема прибавленного титранта в кулонометрическом титровании измеряют время т и ток i электролиза. Процесс образования вещества T в кулонометрической ячейке во время электролиза называется генерацией титранта.
Кулонометрическое титрование при постоянном токе
При кулонометрическом титровании в амперостатическом режиме (при постоянном токе) измеряют времяв течение которого проводился электролиз, и количество электричества Q, израсходованное при электролизе, рассчитывают по формуле (2), после чего находят массу определяемого вещества X по соотношению (1).
Так, например, стандартизацию раствора хлороводородной кислоты методом кулонометрического титрования проводят путем титрования ионов водорода  стандартизуемого раствора, содержащего HCl, электрогенерируемыми на платиновом катоде гидроксид-ионами OH - при электролизе воды:
стандартизуемого раствора, содержащего HCl, электрогенерируемыми на платиновом катоде гидроксид-ионами OH - при электролизе воды:
Образовавшийся титрант - гидроксид-ионы - реагирует с ионами  в растворе:
в растворе:

Титрование ведут в присутствии индикатора фенолфталеина и прекращают при появлении светло-розовой окраски раствора.
Зная величину постоянного токав амперах) и время(в секундах), затраченное на титрование, рассчитывают по формуле (2) количество электричества Q (в кулонах) и по формуле (1) - массу (в граммах) прореагировавшей HCl, содержавшуюся в аликвоте стандартизуемого раствора HCl, внесенного в кулонометрическую ячейку (в генераторный сосуд).
На рис. 3-12 схематически показан один из вариантов электрохимической ячейки для кулонометрического титрования с визуальной (по изменению окраски индикатора) индикацией окончания титрования, с генераторным катодом и вспомогательным анодом.
Генераторный платиновый электрод 1 (в рассматриваемом случае - анод) и вспомогательный платиновый электрод 2 (в рассматриваемом случае - катод) помещены соответственно в генерационный (генераторный) сосуд 3 и вспомогательный сосуд 4. Генерационный сосуд 3 заполнен испытуемым раствором, содержащим определяемое вещество X, фоновый электролит с вспомогательным электроактивным веществом и индикатором. Вспомогательное вещество и само может играть роль фонового электролита; в таких случаях нет необходимости вводить в раствор другой фоновый электролит.
Генерационный и вспомогательный сосуды соединены электролитическим (солевым) мостиком 5, заполненным сильным индифферентным электролитом для обеспечения электрического контакта между электродами. Концы трубки электролитического мостика закрыты пробками из фильтровальной бумаги. В генерационном сосуде имеется магнитный стержень 6 для перемешивания раствора посредством магнитной мешалки.
Электрохимическая ячейка включается в электрическую цепь установки для кулонометрического титрования, способную поддерживать ток постоянным и требуемой величины (например, используют универсальный источник питания типа лабораторного прибора УИП-1 и подобную аппаратуру).
До кулонометрического титрования электроды тщательно промывают дистиллированной водой, в генерационный сосуд вносят раствор с вспомогательным электроактивным (в данных условиях) веществом, при необходимости - фоновый электролит и индикатор.
Поскольку приготовленный таким путем фоновый раствор может содержать электровосстанавливающиеся или электроокисляющиеся примеси, то вначале проводят предэлектролиз фонового раствора в целях электровосстановления или электроокисления примесей. Для этого замыкают электрическую цепь установки и ведут электролиз в течение
некоторого (обычно небольшого) времени до изменения окраски индикатора, после чего цепь размыкают.

Рис. 3-12. Схема электрохимической ячейки для кулонометрического титрования с визуальной индикаторной фиксацией окончания титрования: 1 - рабочий генераторный платиновый электрод; 2 - вспомогательный платиновый электрод; 3 - генерационный сосуд с испытуемым раствором; 4 - вспомогательный сосуд с раствором сильного индифферентного электролита; 5 - электролитический мостик; 6 - стержень магнитной мешалки
После завершения предэлектролиза в генерационный сосуд вносят точно измеренный объем анализируемого раствора, включают магнитную мешалку, замыкают электрическую цепь установки, одновременно включая секундомер, и ведут электролиз при постоянном токе до момента резкого изменения окраски индикатора (раствора), когда сразу же останавливают секундомер и размыкают электрическую цепь установки.
Если анализируемый раствор, вводимый в кулонометрическую ячейку для титрования, содержит примеси электровосстанавливающихся или электроокисляющихся веществ, на превращения которых затрачивается при электролизе некоторое количество электричества, то после предэлектролиза (до прибавления в ячейку анализируемого раствора) проводят холостое титрование, вводя в кулонометрическую ячейку вместо анализируемого раствора точно такой же объем раствора, который содержит все те же вещества и в тех же количествах, что и прибавленный анализируемый раствор, за исключением определяемого вещества X. В простейшем случае к фоновому раствору прибавляют дистиллированную воду в объеме, равном объему аликвоты анализируемого раствора с определяемым веществом.
Время, затраченное на холостое титрование, в дальнейшем вычитают из времени, затраченного на титрование испытуемого раствора с определяемым веществом.
Условия проведения кулонометрического титрования. Должны обеспечить 100% выход по току. Для этого необходимо выполнять, по крайней мере, следующие требования.
1. Вспомогательный реагент, из которого на рабочем электроде генерируется титрант, должен присутствовать в растворе в большом избытке по отношению к определяемому веществу (~1000-кратный избыток). В этих условиях обычно устраняются побочные электрохимические реакции, основная из которых - окисление или восстановление фонового электролита, например, ионов водорода:

2. Величина постоянного тока i = const при проведении электролиза должна быть меньше величины диффузионного тока вспомогательного реагента во избежание протекания реакции с участием ионов фонового электролита.
3. Необходимо как можно точнее определять количество электричества, израсходованное при проведении электролиза, для чего требуется точно фиксировать начало и конец отсчета времени и величину тока электролиза.
Индикация конца титрования. При кулонометрическом титровании ТЭ определяют либо визуальным индикаторным, либо инструментальными (спектрофотометрическими, электрохимическими) методами.
Например, при титровании раствора тиосульфата натрия электрогенерированным йодом в кулонометрическую ячейку прибавляют индикатор - раствор крахмала. После достижения ТЭ, когда в растворе оттитрованы все тиосульфат-ионы, первая же порция электрогенерированного йода окрашивает раствор в синий цвет. Электролиз прерывают.
При электрохимической индикации ТЭ в испытуемый раствор (в генерационный сосуд) помещают еще пару электродов, входящих в дополнительную индикаторную электрическую цепь. Окончание титрования можно фиксировать с помощью дополнительной индикаторной электрической цепи потенциометрически (рН-метрически) или биамперометрически.
При биамперометрической индикации ТЭ строят кривые титрования в координатахизмеряя ток i в дополнительной инди-
каторной электрической цепи как функцию времениэлектролиза в кулонометрической ячейке.
Кулонометрическое титрование при постоянном потенциале
Потенциостатический режим в кулонометрическом титровании используется реже.
Кулонометрическое титрование в потенциостатическом режиме ведут при постоянном значении потенциала, соответствующем потенциалу разряда вещества на рабочем электроде, например, при катодном восстановлении катионов металлов M n + на платиновом рабочем электроде. По мере протекания реакции потенциал остается постоянным до тех пор, пока прореагируют все катионы металла, после чего он резко уменьшается, поскольку в растворе уже нет потенциалопределяющих катионов металла.
Применение кулонометрического титрования. В кулонометрическом титровании можно использовать все типы реакций титриметрического анализа: кислотно-основные, окислительно-восстановительные, осадительные, реакции комплексообразования.
Малые количества кислот (до ~10 -4 -10 -5 моль/л) можно определять кулонометрическим кислотно-основным титрованием электрогенерированными -ионами, образующимися при электролизе воды на катоде:
Можно титровать и основания ионами водорода генерируемыми на аноде при электролизе воды:

При окислительно-восстановительном бромометрическом кулонометрическом титровании можно определять соединения мышьяка(III), сурьмы(III), иодиды, гидразин, фенолы и другие органические вещества. В роли титранта выступает электрогенерируемый на аноде бром:

Осадительным кулонометрическим титрованием можно определять галогенид-ионы и органические серосодержащие соединения электрогенерированными катионами серебра катионы цинка - электрогенерированными ферроцианид-ионами и т.д.
Комплексонометрическое кулонометрическое титрование катионов металлов можно проводить анионами ЭДТА, электрогенерированными на катоде из комплексоната ртути(II).
Кулонометрическое титрование обладает высокой точностью, широким диапазоном применения в количественном анализе, позволяет определять малые количества веществ, малостойкие соединения (поскольку они вступают в реакции сразу же после их образования), например, меди(I), серебра(II), олова(II), титана(III), марганца(III), хлора, брома и др.
К достоинствам метода относится также и то, что не требуется приготовления, стандартизации и хранения титранта, так как он непрерывно образуется при электролизе и сразу же расходуется в реакции с определяемым веществом.
Цели изучения темы
На основе знания теоретических основ метода кулонометрического титрования и выработки практических умений научиться обоснованно выбирать и практически применять данный метод анализа для количественного определения вещества; уметь проводить статистическую оценку результатов кулонометрического титрования.
Целевые задачи
1. Научиться проводить количественное определение массы натрия тиосульфата в растворе методом кулонометрического титрования.
2. Научиться проводить стандартизацию раствора хлороводородной кислоты методом кулонометрического титрования.
3. Решение типовых расчетных задач.
На изучение темы отводится одно лабораторное занятие из двух, описанных в данном пособии. Рекомендуется проводить лабораторную работу «Определение массы натрия тиосульфата в растворе методом кулонометрического титрования».
Задание для самоподготовки
К занятию необходимо знать
1. Принцип методов кулонометрии.
2. Сущность метода кулонометрического титрования при определении:
а) натрия тиосульфата;
б) хлороводородной кислоты.
Необходимо уметь
1. Писать уравнения электрохимических реакций, протекающих на электродах при кулонометрическом титровании:
а) натрия тиосульфата;
б) хлороводородной кислоты.
2. Писать уравнения электрохимических реакций, протекающих в растворе при кулонометрическом титровании:
а) натрия тиосульфата;
б) хлороводородной кислоты.
3. Рассчитывать количество электричества и массу (концентрацию) вещества по результатам кулонометрического титрования.
4. Обрабатывать результаты параллельных определений вещества методом математической статистики.
Список литературы
1.Учебник. - Книга 2, глава 10. - С. 481-492; 507-509; 512-513.
2.Харитонов Ю.Я., Григорьева В.Ю. Примеры и задачи по аналитической химии.- М.: ГЭОТАР-Медиа, 2009.- С. 240-244; 261-264; 277-281.